
|
|||||
|
CASE REPORT / CAS CLINIQUE
ADRENOLEUCODYSTROPHIE LIEE A L’X OBSERVE A COTONOU (BENIN)
X-LINKED ADRENOLEUKODYSTROPHY IN COTONOU. A CASE REPORT.
E-Mail Contact - AVODE Dossou Gilbert :
avogil15@gmail.com
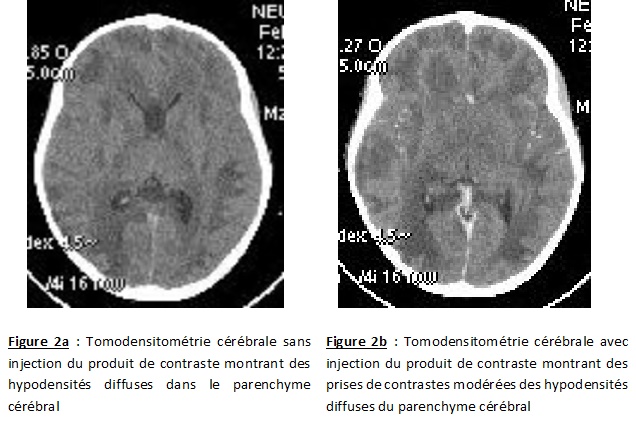
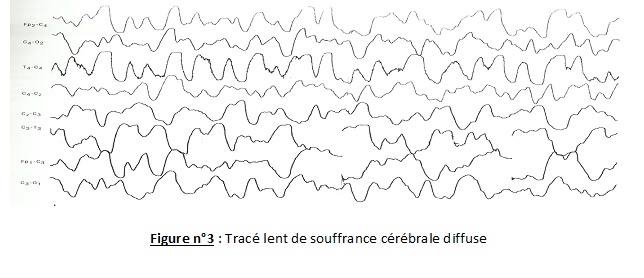
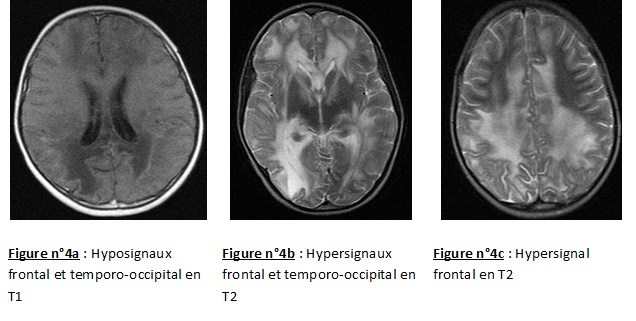
RESUME L’adrénoleucodystrophie (ALD) liée à l’X est une maladie génétique liée au chromosome sexuel X. Les auteurs rapportent le premier cas révélé par des crises épileptiques partielles myocloniques et observé au Bénin. Le diagnostic était fait sur la base d’une démyélinisation du système nerveux central, une accumulation des acides gras à très longue chaîne (AGTLC) et une mutation dans le gène ABCD1. ABSTRACT Adrenoleukodystrophy (ALD) is an X-linked genetic disease related to sex chromosome X. The authors report the first case revealed by partial seizures and myoclonic observed in Benin. The diagnosis was made on the basis of a demyelination of the central nervous system, an accumulation of very long chain fatty acids (VLCFA) and a mutation in the gene ABCD1. INTRODUCTION L’adrénoleucodystrophie (ALD) liée à l’X est une maladie génétique liée au chromosome sexuel X et qui associe une démyélinisation du système nerveux central et/ou périphérique, une insuffisance surrénalienne et une accumulation des acides gras à très longue chaîne (AGTLC, acides gras ayant un nombre de carbones supérieur à 22) [2]. Son incidence est de 1/17 000 naissances [4]. Plusieurs cas ont fait l’objet de description au Maghreb et en Occident [ 3, 5, 6 ], aucun n’a été décrit au Bénin. C’est pourquoi les auteurs rapportent le premier cas d’ALD révélé par des crises épileptiques partielles myocloniques observé au BENIN. OBSERVATION Enfant A.R. âgé de 6 ans, écolier au cours préparatoire (CP), est admis dans le service de Pédiatrie et de génétique médicale pour des crises épileptiques sub-intrantes et des céphalées le 14 Février 2014. En effet, en Octobre 2013, l’enfant a présenté des difficultés inattendues au CP alors que la maternelle et le cours d’initiation (CI) s’étaient bien déroulés. Il était agité, avait des difficultés à se concentrer. En novembre 2013, on notait un strabisme de l’il droit avec baisse de l’acuité visuelle. En Janvier 2014, les maîtres de son école ont constaté deux épisodes d’ictus mnésique où l’enfant se perdait et n’arrivait pas à reconnaitre sa place dans la classe. Des épisodes d’absences récurrents, une boulimie ont été signalés et il fut mis sous Gardenal 50mg par son Médecin traitant avec la demande d’une consultation neurologique. Devant la survenue de céphalées atroces, de vomissements en jet, de clonies du membre thoracique droit dans un contexte fébrile, l’enfant fut hospitalisé le 14 Février 2014. Dans ses antécédents, on retrouve une notion de trait de drépanocytose AS. Il est 3ème d’une fratrie de 3 enfants (figure n°1). L’examen clinique à l’entrée a retrouvé une température à 38°5, une tension artérielle à 112/67mmHg, un bon état de conscience et une agitation psychomotrice. Devant le syndrome de réponse inflammatoire systémique, le syndrome d’hypertension intracrânienne et le syndrome d’irritation corticale, l’hypothèse d’une encéphalite fut évoquée. La tomodensitométrie cérébrale réalisée en urgence objective des hypodensités diffuses prenant partiellement le contraste (figure n°2a et b) et faisant évoquer une pan-encéphalite. L’enfant a été mis sous une triple antibiothérapie à base de Ciprofloxacine (400mg/j), Flagyl (750mg/j), et Ceftriaxone (1g/j) à laquelle avait été associé le Bêtaméthasone (40mg/j), et le Phénobarbital (50mg/j). L’électroencéphalogramme était globalement lent pour l’âge (figure n°3). Le fond d’il montrait un dème papillaire bilatéral. La biologie standard était sans particularité. Les sérologies HIV et toxoplasmique étaient négatives. L’évolution sous le traitement une semaine après, était marquée par la régression de la fièvre, mais les crises épileptiques partielles myocloniques du membre thoracique droit persistaient. Une consultation neurologique fut demandée et retrouve un état d’agitation, agressivité, cris, manifestations d’opposition sans frustration identifiée, une baisse de l’acuité visuelle, une reconnaissance des parents par l’odorat, une fluctuation dans la compréhension et dans l’audition, une tendance à parler le dialecte alors qu’il comprenait et parlait très bien le français, un fauchage à la marche, des troubles de l’oculomotricité avec un strabisme divergent de l’il gauche mais une conservation des réflexes photomoteurs qui sont symétriques sans asymétrie pupillaire, un syndrome tétrapyramidal constitué d’une trépidation épileptoïde bilatéral sans Rossolimo, des réflexes ostéo-tendineux vifs de façon bilatérale, un Hoffmann à gauche et 3 épisodes d’absences cliniques avec révulsion oculaire et arrêt de l’activité de quelques secondes. Le Valproate de Sodium 200mg deux fois par jour, a été introduit. Mais devant ces importantes hypodensités sous corticales, nous avons évoqué une leucodystrophie et l’IRM cérébrale du 20 Février 2014 a objectivé un hyposignal T1, un hypersignal T2 aux lobes frontaux et occipitaux faisant évoquer une leucodystrophie de type adrénoleucodystrophie d’origine héréditaire (figure n°4). Les parents prennent en Avril 2014 à Paris un 2ème avis. Le dosage des acides gras à très longues chaînes (AGTLC) réalisé montrait un profil très anormale et évoquait une maladie peroxysomale de type adrénoleucodystrophie liée à l’X. Le bilan endocrinien, notamment l’ACTH était à 10 fois la normale. Le cortisol matinal était normal. Sur le plan génétique, on notait une mutation dans le gène ABCD1 aussi bien chez l’enfant que chez sa mère. Aucun traitement spécifique n’a été introduit du fait de l’installation des troubles neurologiques. Il fut conseillé à la famille de dépister les frères ainés de l’enfant A.R, et institué dans le cas échéant, une biothérapie avant l’installation des troubles neurologiques. En Mai 2015, soit plus d’un an après, l’enfant est alité, présente une cécité bilatérale, une perte totale du langage et une quadriplégie. Les crises d’épilepsie ont largement diminué sous Valproate de sodium 200mg 1cp matin et soir.  Figure 1
DISCUSSION L’ALD est une maladie génétique récessive liée à l’X qui atteint principalement la substance blanche cérébrale, les corticosurrénales et les testicules. Elle est à l’origine d’un déficit de catabolisme dans le peroxysome des acides gras à très longue chaîne [1]. Plusieurs phénotypes sont décrits. La forme la plus fréquente est la forme cérébrale de l’enfant qui représente environ la moitié des cas. Elle apparait après un développement psychomoteur normal. La dégradation neurologique cérébrale est rapidement progressive conduisant à un état végétatif en deux ans [2, 6]. C’est le cas de notre observation où avant l’âge de 5 ans, l’enfant a connu un bon développement psychomoteur avec acquisition de la marche à un an, le langage à 2 ans. Les troubles neurologiques sont apparus après l’âge de 5 ans et ont évolué de façon rapidement progressive en 10 mois, aboutissant à une perte d’autonomie complète, une perte du langage et une cécité bilatérale. La seconde forme est l’adrénomyéloneuropthie de l’adulte (25 %), secondaire à la démyélinisation des cordons postérieurs et antérieurs de la moelle avec respect de la substance blanche cérébrale. Les manifestations neurologiques apparaissent en moyenne autour de 30 ans sous la forme d’une paraparésie spastique progressive et d’une neuropathie périphérique modérée [8, 9]. Les autres formes, plus rares, sont l’atteinte cérébrale de l’adolescent (5 %), de l’adulte (3 %) et une insuffisance surrénalienne isolée (10 %) [4]. Sur le plan biochmique, l’ALD se caractérise par une accumulation d’AGTLC due à un déficit de leur bêta-oxydation dans le péroxysome [6, 10]. Le gène de l’ALD, localisé en Xq28, code pour un hémi-transporteur (appelé ALDP) localisé dans la membrane du péroxysome [4]. Soixante-seize mutations ont été décrites dans la littérature, la même mutation pouvant être associée à plusieurs phénotypes différents. Il n’y a pas de corrélation entre le type de la mutation et le phénotype [4, 9]. Dans notre cas, l’enfant et sa mère sont porteurs de la mutation ABCD1. Le diagnostic repose sur la mise en évidence d’une augmentation des AGTLC dans le plasma ou les fibroblastes Sur le plan thérapeutique, la greffe allogénique de moelle osseuse est à ce jour l’unique traitement à condition qu’il soit institué tôt, en l’absence des lésions neurologique [7]. Une IRM et des explorations neuropsychologiques sont répétées tous les six mois chez les malades asymptomatiques afin de les transplanter s’il existe un donneur compatible dès les signes précoces d’atteinte cérébrale [8]. Il existe donc un intérêt à faire un dépistage précoce. C’est ce qui a d’ailleurs été proposé à la famille afin de savoir si les frères ainés de A.R sont porteurs de la mutation. Dans le cas échéant, et étant encore asymptomatiques, on pourrait leur proposer une greffe allogénique. L’insuffisance surrénalienne est traitée par une corticothérapie. Dans notre observation, en dépit de l’augmentation de l’ACTH qui est supérieur à 10 fois la normale, il n’existait pas d’insuffisance surrénalienne. CONCLUSION L’ALD est une affection rare mais qui existe dans notre milieu de travail. Il est important d’y penser devant un tableau de pan-encéphalite et faire réaliser une IRM cérébrale. La confirmation du diagnostic implique le dosage des AGTLC et la recherche de la mutation génétique ABCD1. Le dépistage précoce de cette affection, permettra la mise en place de la greffe allogénique pouvant influer sensiblement sur l’apparition des lésions neurologiques.
REFERENCES
|
© 2002-2018 African Journal of Neurological Sciences.
All rights reserved. Terms of use.
Tous droits réservés. Termes d'Utilisation.
ISSN: 1992-2647