CASE REPORT / CAS CLINIQUE
CEREBROTENDINOUS XANTHOMATOSIS : DESCRIPTION OF A CASE
CEREBROTENDINOUS XANTHOMATOSIS : A PROPOS D'UN CAS
- Neurology Department, SSK Goztepe Hastanesi Göztepe ISTANBUL/TURKEY
E-Mail Contact - ISIK Nihal :
ABSTRACT
A 24 year old woman was referred for evaluation of progressive dementia and abnormal gait. She presented with mental impairment and a spastic gait. Physical examination showed noduler thickening of both achilles tendons and high arched feet. Her parents recognized this thickness when she was 7 years old. Neurologic examination showed mild spasticity in the lower extremities, normal power, diffuse hyperreflexia, moderate truncal ataxia, mild lower extremity ataxia, stocking distribution sensory loss to light touch and loss of proprioception in the legs. Neurophyschologic tests showed widespread neurocognitive deficits with poor attention and concentration and seriously impaired memory.
The association of this neurological and physical findings and neuroradiological and laboratory examinations correspond to a rare entity named Cerebrotendinous Xanthomatosis.
RESUME
La xanthomatose cérébro tendineuse est une maladie héréditaire rare caractérisée par des taux de cholestérol élevés au niveau du plasma et du cerveau. Nous rapportant le cas d’une patiente de 24 ans, hospitalisée pour une démence progressive et des troubles de la marche.
Keywords: cerebrotendinous, xanthomatosis
INTRODUCTION
Cerebrotendinous Xanthomatosis is a rare hereditary progressive storage disease characterized by marked increases in plasma and brain cholestanol levels. Clinical symptoms may be evident in differrent organ systems but are usually dominated by the classical triad of the disease: Juvenile cataracts, tendon xanthomas and progressive neurological impairment (18). The diagnosis can be made biochemically, genetically and radiologically. Early diagnosis is imperative in patients with Cerebrotendinous Xanthomatosis as the pharmalogical treatment has been shown to slow or even reverse the progression of the disease (20).
REPORT OF CASE
A 24-year-old woman was referred for evaluation of progressive dementia and abnormal gait. Her gestation, birth and early development were normal, except she had intractable diarrhea during childhood. She attended school education only up to fifth class of primary school and left the school because of learning problems. Her parents recognized that she had mental insuffiency, poor attention and concentration with impaired memory and by the time passing these findings became more evident. Also noduler thickness on her achilles tendons were recognized by her family when she was 7 years old, and they became larger within the years. Their colour was grayish yellow and they were painless. Cataracts were found at age 10 and her walking became unsteady at age 22. There was no family history of neurological or systemic illness.
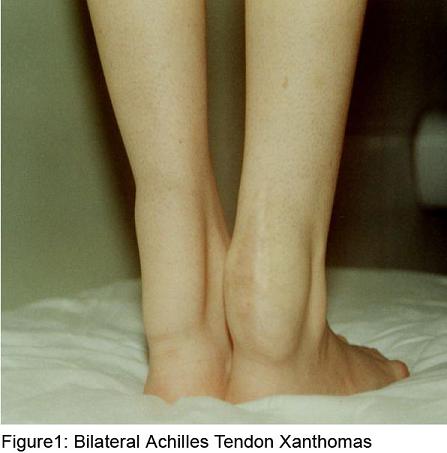
Xanthomas on the achilles tendons and high arched feet were detected on her physical examination (figure 1). Her neurological examination showed mental insuffiency with subnormal Mini-Mental Status Score (22/30), mild spasticity in the lower extremities, normal power, diffuse hyperreflexia, moderate truncal ataxia, stocking distribution sensory loss to light touch and loss of proprioception in the legs and impaired heel toe walking. Neurophyschologic tests showed widespread neurocognitive deficits with poor attention and concentration and seriously impaired memory.
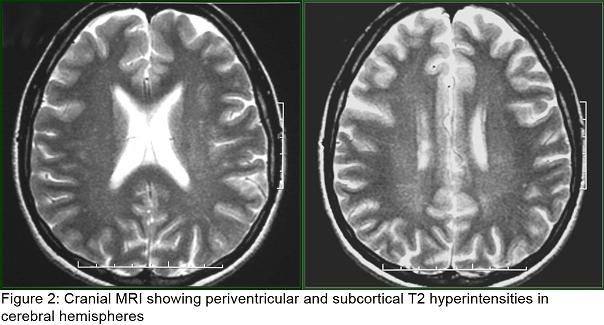
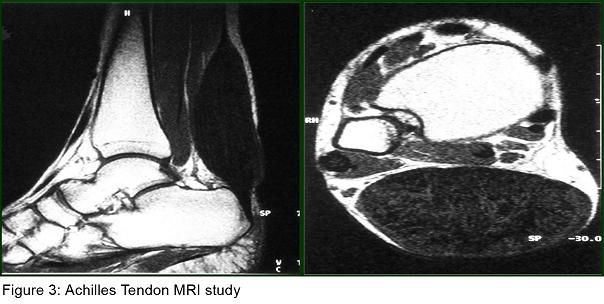
Laboratory tests revealed normal total plasma cholesterol, LDL cholesterol and triglycerides but elevated plasma cholestanol level (14 mg/L ) ( normal, 2 to 6 mg/L). Cardiological examination including electrocardiography and echocardiography were normal. Her abdominal USG and CT and thorax CT were normal. Her EEG was normal. Sensorymotor neuropathy was detected in her electrophysiological examination, including nerve conduction study and electromyography (EMG). Somatosensorial evoked potential studies revealed prolonged P40 responses with posterior tibial nerve stimulation; brainstem auditory evoked potentials and visual evoked potentials were normal. Cranial MRI showed periventricular and subcortical T2 hyperintensities in cerebral hemispheres (figure2). Her MR Spectroscopy was reported as normal. MRI study of achilles tendons showed a diffuse enlargement of the tendon from musculotendinous junction to calcaneal insertio, and also showed heterogen hypointensity signal on T1 and T2 images. Cystic degeneration area in the right achill tendon was named as xanthoma (figure3).
With these physical, radiological and biochemical findings the patient was diagnosed as Cerebrotendinous Xanthomatosis.
Chenodeoxycholic acid was put on for the treatment. Her gait had been improved with the treatment but it did not provide any clinical benefit especially including intellectual functions.
DISCUSSION
Cerebrotendinous Xanthomatosis is a rare autosomal recessive lipid storage disease with prominent neurological features. The disease was first described by Van Bogaert at el. in 1937 (14).
The disease is associated with mutations in CYP 27, which encodes mitochondrial sterol 27-hydroxylase, an enzyme that catalyses the oxidation of sterol intermediates during bile acid synthesis (10,11). The loss of this enzyme results in accumulation of cholestanol in many tissues, particularly in the brain. How specifically cholestanol accumulation produces functional abnormalities is unknown and should be investigated further. As a result, it leads to formation of multıple xanthomas of tendons and other tissues, dementia, premature cataracts, progressive cerebellar ataxia, spinal cord paresis, peripheral neuropathy and chronic diarrhea (8). It may present with chronic diarrhea and bilateral cataracts in early childhood. Patients usually develop tendon xanthomas and neurologic symptoms after second decade of life. Achilles tendon is the most common site of tendon xanthomas, but quadriceps, triceps and finger extensor tendons can be involved also. Altough they have the same appearance as xanthomas found in patients with familial hypercholesterolemia or hyperlipoproteinemia, biochemical analysis reveals that they contain high amounts of cholestanol and little cholesterol (6). Biopsy of the tendon xanthomas is rarely necessary for diagnosis. Our patient presented almost all of the symptoms of the disease. She had intractable diarrhea during childhood. Cataracts were found at age 10. Xanthomas were recognised when she was 7 years old. She had learning problems and was unsuccessful at the school. The clinical pattern was characterized by progressive dementia and abnormal gait.
Onset of the disease usually occurs toward the end of the first decade of life, and most individuals live beyond middle age (8). Early onset cataracts are characteristic of Cerebrotendinous Xanthomatosis. But other ophthalmological findings have occasionally been reported. Besides cataracts, the second most frequent ocular abnormality is paleness of the optic disc (4). Cataracts were found in our patient at age of 10, no other abnormality was observed in her ophtalmological examination.
Peripheral neuropathy is a prominent feature of Cerebrotendinous Xanthomatosis as evidenced by pes cavus deformities, loss of deep tendon reflexes and loss of vibration perception (9). Stocking distribution sensory loss to light touch and loss of proprioception in the legs were detected on our patient’s neurological examination and her electrophysiological examination included nerve conduction study and electromyography (EMG) was found compatible with sensorymotor neuropathy. In CTX, electrophysiological studies show prolonged central conduction times in short latency somatosensory evoked potentials with tibial nerve stimulation but normal conduction velocities with median nerve stimulation. Brainstem auditory evoked potentials and visual evoked potentials are abnormal in approximately one-half of patients (9). Our patient’s somatosensory evoked potential studies revealed prolonged P40 responses with posterior tibial nerve stimulation; brainstem auditory evoked potentials and visual evoked potentials were normal.
Seizures are encountered in 40-50% and can be presenting symptom (8). As seen in our patient during childhood period intractable diarrhea can be a nearly major manifestation(16,19). She did not have any epileptic seizure. However her cardiological examinations including echocardiography and electrocardiography were normal, premature atherosclerosis and cardiac complications have been reported among the systemic manifestations of Cerebrotendinous Xanthomatosis. In some cases, myocardial infarction has been the cause of sudden death. This suggests that careful cardiac examination should be considered, even in the absence of clinical manifestations (5).
Biochemical changes are very important for the diagnosis of CTX. The cholesterol and triglycerides concentration in serum are normal but cholestanol levels in serum and erythrocytes are elevated. Levels of cholestanol are particularly high in bile, xanthomas and brain. Bile acid production is decreased markedly, which leads to reduce chenodeoxycholic acid concentration in bile. Excretion of bile acid precursors is increased in bile and urine. Serum cholesterol levels are typically not elevated in Cerebrotendinous Xanthomatosis (20,10,13).
Also neuroradiological studies can be used for the diagnosis of CTX. Cerebral CT reveals the presence of hypodense nodules in the cerebellum and diffuse white matter hypodensity. Magnetic resonance imaging findings typically include bilateral and almost symmetrical increase of the signal intensity on the T2-weighted images in the cerebellar and periventricular cerebral white matter, the basal ganglia, the dentate nuclei and the brainstem as well as cerebellar and cerebral atrophy (17). However, this finding has been seen inconsistently on conventional MRI (1). The dynamic of the CNS pathology in Cerebrotendinous Xanthmatosis is complex and whether demyelination or axonopathy has primary importance in the pathogenesis of cerebrotendinous Xanthomatosis pathology is not known (1). Also, Proton MR Spectroscopy data demonstrates widespread axonal damage (decrease in N-acetylaspartate) and diffuse brain mitochondrial dysfunction (increase in brain parenchymal lactate) (7). Periventricular and subcortical T2 hyperintensities were found in our patient’s cranial MRI. Her MR Spectroscopy was reported as normal.
In CTX, MR study of achilles tendon shows a diffuse enlargement of the tendon with multiple areas of signal changes on T1 and T2 demonstratative of the lipid deposits, interpossed between the isosignal zones, they may correspond to the inflammatory reaction secondary to the accumulation of cholesterol and cholestanol (12). Our patient’s achilles tendon MR study showed heterogen hypointensity signal on T1 and T2 images and cystic degeneration was named as xanthoma. This was also compatible with CTX.
Early diagnosis is imperative in CTX, as the pharmalogical treatment with chenodeoxycholic acid ( or with a combination of chenodeoxycholic acid and HMG-CoA reductase inhibitors ) has been shown to slow or even reverse the progression of the disease. Treatment with chenodeoxycholic acid has been reported to be beneficial in some patients who showed a correction of biochemical abnormalities, a reversal of neurological symptoms, and an improvement in somatosensory evoked potentials and in the MRI scan (15).
Also simvastatin, a HMG Co A reductase inhibitor was found to be effective in decreasing serum cholestanol concentrations alone, but greater reductions were achieved after the addition of the bile acid chenodeoxycholic acid; suggesting a synergistic effect of this combination (2,3).
We used chenodeoxycholic acid for the treatment and minimal improvement of gait pattern was observed but no improvement was found in her intellectual functions. But the long term clinical outcome warrents further evaluation. We decided to follow up the patient with this treatment.
CONCLUSION
CTX is a rare progressive lipid storage disease with prominent neurological features. Physicians should be aware of possible clinical, physical manifestations and neurological findings; because early diagnosis and treatment may reduce the risk of long term neurologic sequelae.
The possibility of early treatment may improve the neurological symptoms of Cerebrotendinous Xanthomatosis underscores the importance of carefull screening and genetic counseling of asymptomatic relatives of patients with this disorder.
REFERENCES
- BARKHOF F, VERRIPS A, WESSELING P, VAN DER KNAAP MS. Cerebrotendinous xanthomatosis: The spectrum of imaging findings and the correlation with neuropathologic findings. Radiology 2000 Dec;217 (3):869-76
- BURNETT JR, MOSES EA., CROFT KD., BROWN AJ.,GRAINGER K. Clinical and biochemical features, molecular diagnosis and long term management of a case of Cerebrotendinous xanthomatosis. Clin. Chim Acta 2001 Apr;306(1-2)63-9
- DONAGHY M., KING RH., MCKERAN RO., SCHWARTZ MS. Cerebrotendinous Xanthomatosis: Clinical, electrophysiological and nerve biopsy findings and response to treatment with chenodeoxycholic acid. J. Neurol 1990 Jun;237 (3):216-9
- DOTTI MT, RUFA A, FEDERICO A. Cerebrotendinous Xanthomatosis: Heterogeneity of clinical phenotype with evidence of previously undescribed ophthalmogical findings J. Inherit Metab Dis 2001 Dec; 24
- DOTTI MT, MONDILLO S , PLEWNIA K, AGRICOLE E, FEDERICO A Cerebrotendinous Xanthomatosis: Evidence of lipomatous hpertrophy of the atrial septum J. Neurol 1998 Nov; 245 (11):723-6
- DE JONG JG, VAN GENT CM, DELLEMAN JM. Cerebrotendinous cholestanolosis in relation to other cerebralxanthomatosis. Clin Neurol Nerosurg 1997;79(4):253-72
- DE STEFANO N, DOTTI MT, MORTILLA M. FEDERICO A. Magnetic resonance imaging and spectroscopic changes in brains of patients with Cerebrotendinous xanthomatosis. Brain 2001 Jan;124 (Pt 1):121-31
- K. F. KO, K. W. LEE: Cerebrotendinous Xanthomatosis in three siblings from a Chinese Family. Singapore Med J 2001 Vol42(1):030-032
- KURTZKY A, BERGINER VM, KORCYZN AD. Peripheral neuropathy in Cerebrotendinous Xanthomatosis. Neurology 1979;29:880-1
- LAMON – FAVA S, SCHAEFER E, GARUTI R, SCALEN G, CALANDRA S. Two novel mutations in the sterol 27 – hydroxylase gene causing Cerebrotendinous Xanthomatosis Clin.Genet. 2002 Mar; 61(39):185-191
- MOHAMMED H MOGHADASIAN, GERALD SALEN, JIRI J. FROHLICH, CHARLES H. SCUDAMORE. Cerebrotendinous Xanthomatosis: A rare disease with diverse Manifestations. Arch Neurol.Apr 2002;59:527-529
- PARDO J, PRIETO JM, RODRIGUEZ JR. VADILLO J. Magnetic resonance of the brain and Achilles tendon in cerebrotendinous xanthomatosis. Neurologia 1993 Oct;8(8):268-70
- SALEN G, TINT GS, ELIAV B, DEERING N, MOSBACH EH. Increased formation of ursodeoxycholic acid patients treated with chenodeoxycholic acid J. Clin Invest 1974 Feb; 53 (2):612-21
- VAN BOGAERT L, SCHERER H, EPSTEIN E. Une Forme Cerebrale de la cholesterionose Generalise Paris: Masson 8 Cie, 1937
- VAN HEIJST AF, VERRIPS A, WEVERS RA. CRUYSBERG JR. RENIER WO. Treatment and follow up of children with Cerebrotendinous Xanthomatosis Eur. J Pediatr 1998 Apr;157 (4):313-6
- VAN HEIJST AF, WEVERS RA, TANGERMAN A, CRUYSBERG JR, RENIER WO, TOLBOOM JJ. Chronic diarrhoea as a dominating symptom in two children with cerebrotendinous xanthomatosis. Acta Pediatr 1996 Aug;85(8):932-6
- VANRIETVELDE F, LEMMERLING M, MESPREUVE M. CREVITS L. MRI of the brain in Cerebrotendinous Xanthomatosis (Van Bogaert- Scherer – Epstein disease) Eur Radiol 2000; 10(4):576-8
- VERRIPS A, HOEFSLOOT LH, STEENBERGEN GC, THEELEN JP, WEVERS RA, GOBREELS FJ, et al. Clinical and molecular genetic characteristics of patients with CTX. Brain 2000; 123: 980-19
- VERRIPS A, VAN ENGELEN BG, WEVERS RA, VAN GEEL BM. Presence of diarrhea and absence of tendon xanthomas in patients with cerebrotendinous xanthomatosis Arch Neurol.2000 Apr;57(4):520 -4
- VERRIPS A, WEVERS RA, VAN ENGELEN BG, KEYSER A, WOLTHERS BG, et al. Effect of simvastatin in addition to chenodeoxycholic acid in patients with cerebroxanthomatosis. Metabolism 1999b; 48: 233-8
|
|
