
|
|
|
CASE REPORT / CAS CLINIQUE
COMPLICATIONS NEUROLOGIQUES DE LA CARENCE EN THIAMINE (VITAMINE B1) : A PROPOS D’UN CAS AVEC REVUE DE LA LITTERATURE.
NEUROLOGICAL COMPLICATIONS OF THIAMINE DEFICIENCY, A CASE REPORT WITH LITERATURE REVIEW.
E-Mail Contact - EL HADDAD Mariyam :
elhaddadmariyam@gmail.com
RÉSUME Introduction La carence en thiamine peut se manifester par deux formes cliniques : une forme cardiaque nommée béribéri humide, et une forme neurologique qui est le béribéri sec ; cette dernière peut se traduire cliniquement par une neuropathie périphérique, ou par une encéphalopathie de Gayet-Wernicke, caractérisée par la triade classique : ophtalmoplégie, ataxie et état confusionnel. Cette encéphalopathie est souvent difficile à identifier et son diagnostic est retardé. Observation Nous rapportons le cas d’une patiente de 27 ans, suivie pour leucémie aigue promyélocytaire réfractaire, avec atteinte du système nerveux central, ayant eu une alimentation parentérale prolongée, en raison des vomissements incoercibles et persistants, suite à laquelle elle a développé une encéphalopathie de Wernicke, ayant bien évolué sous supplémentation vitaminique intra-veineuse. Commentaires Nous aborderons dans ce travail, l’intérêt du diagnostic précoce, les différents contextes évocateurs et les aspects clinique, radiologique ainsi que les options thérapeutiques dans l’encéphalopathie de Wernicke. Mots-clés: Déficit en thiamine, Encéphalopathie de Wernicke, Leucémie, Nutrition parentérale. ABSTRACT Background Thiamine deficiency can manifest itself in two clinical forms: a cardiac form called wet beriberi, and a neurological form which is dry beriberi; the latter can result clinically by peripheral neuropathy, or by Gayet-Wernicke encephalopathy, characterized by the classic triad: ophthalmoplegia, ataxia and confusional state. This encephalopathy is often difficult to identify and its diagnosis is delayed. Observation We report the case of a 27-year-old patient, followed for refractory acute promyelocytic leukemia, with central nervous system damage, having had a prolonged parenteral diet, due to incoercible and persistent vomiting, following which she developed an encephalopathy of Wernicke, having evolved well under intravenous vitamin supplementation. Comments In this work, we will discuss the interest of early diagnosis, the different suggestive contexts and the clinical, radiological aspects as well as the therapeutic options in Wernicke’s encephalopathy. Key Words: Leukemia, Parenteral nutrition, Thiamine deficiency, Wernicke’s encephalopathy. INTRODUCTION La thiamine (ou vitamine B1) est une vitamine hydrosoluble qui passe la barrière hémato-encéphalique, d’origine uniquement alimentaire (pas de synthèse endogène de la thiamine), elle est présente dans presque toutes les viandes (volaille, porc…), céréales (riz, son…), légumes et légumineuses. Elle est absorbée par le duodénum et est stockée principalement dans le foie mais également au niveau du cœur et des muscles. Les besoins journaliers sont de 1,1 à 1,2 mg/j, et en cas de déficit, les troubles apparaissent au bout de 2 à 3 semaines. La thiamine est la forme active de la thiamine pyrophosphate ou thiamine diphosphate, elle représente un cofacteur des pyruvates déshydrogénase, de l’acide alpha-ketoglutarique, et de la trans-ketolase ; ce sont trois enzymes clés du métabolisme des carbohydrates. Le pyruvate déshydrogénase catalyse la conversion du pyruvate à l’acétyl-CoA. Ce métabolisme nécessite la présence de la thiamine pour empêcher l’accumulation des lactates (7). Le cycle de l’acide citrique est une voie métabolique centrale impliquée dans la régulation des glucides, des lipides et le métabolisme des acides aminés et sa carence ou son déficit complet, génère une accumulation de l’acide lactique, et produit une acidose lactique très sévère en inhibant la production de nombreuses molécules, y compris les neurotransmetteurs acide glutamique et GABA. La thiamine peut également participer directement à la neuromodulation (14,23). La thiamine dans le corps humain a une demi-vie de 18 jours et est rapidement épuisée, en particulier lorsque la demande métabolique dépasse l’apport. Le déficit en thiamine peut être en rapport avec une alimentation inadéquate, une baisse de l’absorption, un défaut d’utilisation, ou un excès d’utilisation, il peut survenir chez les patients sévèrement atteints, en cas d’augmentation du métabolisme du glucose (ex. choc septique, postopératoire…), en cas de malnutrition prolongée (ex : syndrome de re-nutrition) ou en cas d’une élimination excessive (ex : atteinte rénale sévère). Dans tous les cas, une supplémentation parentérale ou entérale est primordiale pour éviter les acidoses lactiques secondaires à la carence en thiamine. L’encéphalopathie de Wernicke a été décrite pour la première fois en 1881 par Carl Wernicke, avec la triade classique : paralysies oculomotrices, troubles de la conscience et ataxie. Il s’agit d’une complication neuropsychiatrique aiguë secondaire à un déficit en thiamine, fréquemment rencontrée chez les grands consommateurs d’alcool. Dans le cas d’un traitement inadéquat, le déficit en thiamine provoque des lésions structurelles cérébrales définitives, qui se manifestent par des troubles mnésiques antérogrades et rétrogrades sévères et souvent irréversibles (25). Actuellement, différents tests mesurant l’activité de la thiamine pyrophosphate (forme active de la thiamine) ont été développés. Ils permettent d’identifier les patients carencés qui sont à risque de développer une encéphalopathie de Wernicke (EW). Ces tests ne sont cependant pas disponibles dans l’urgence. Le diagnostic d’EW doit être posé de manière présomptive afin de traiter les patients aussi rapidement que possible (4). CAS CLINIQUE Mme M.L., âgée de 27ans, sans antécédents médico-chirurgicaux ni alcoolisme notables, suivie depuis Août 2014 pour une leucémie aigue promyelocytaire (LAM3), avec au caryotype une trisomie du chromosome 8 (15,17) et un gène FLT3 muté. Elle a été initialement traitée selon le protocole APL 2006. L’induction a été compliquée d’une pancréatite stade B, nécessitant l’arrêt du Vesanoid avec un relais par l’Arsenic. Ayant été réfractaire à cette première ligne de chimiothérapie, un rattrapage par Idarubicine et Trisenox a permis l’obtention d’une première rémission complète moléculaire 3 mois après le début du traitement, consolidé par 3 cycles d’Arsenic, puis un entretien par Purinethol 90mg/m²/semaine associé au méthotrexate15mg/m² /semaine par voie orale. Une rechute moléculaire a été observée 2 mois après le début du traitement d’entretien. Par la suite, elle a reçu une seconde chimiothérapie de rattrapage par Gemtuzumab-Ozogamycine (Mylotarg) associé au trioxide d’Arsenic, suivie d’une allogreffe géno-identique en 2ème rémission complète. La patiente a été conditionnée par Cyclophosphamide, associé au Busulfan. Le processus a été compliqué d’une GVH (réaction du greffon contre l’hôte) digestive grade 2 cortico-sensible. Une deuxième rechute moléculaire a été observée à 5 mois de l’allogreffe, nécessitant l’arrêt de la ciclosporine et des corticoïdes, et une perfusion de DLI (Donnor Lymphocytes Infusion). La patiente a rechuté en neuro-méningé, et a donc reçu une nouvelle induction par Gemtuzumab Ozogamycine (Mylotarg) associé au trioxide d’Arsenic et à 12 ponctions lombaires thérapeutiques. Le traitement de rattrapage s’était compliqué d’une aplasie profonde prolongée, dans un contexte de vomissements incoercibles rebelles aux traitements usuels, suite auxquels elle avait été mise sous nutrition parentérale exclusive et prolongée pendant huit semaines, sans supplémentation vitaminique. A la 6ème semaine d’hospitalisation, la patiente a présenté de façon brutale un trouble oculomoteur, à type de nystagmus vertical bilatéral, avec flou visuel bilatéral, associé à un syndrome cérébelleux cinétique qui s’est manifesté par un élargissement de la surface de sustentation, une marche avec les bras écartés, une hypermétrie, une dyschronométrie, avec des réflexes rotuliens pendulaires bilatéraux à l’examen clinique, sans déficit sensitif ni moteur. Une TDM cérébrale réalisée est revenue normale. Une IRM cérébrale faite a mis en évidence une lésion bithalamique ventrale et postérieure en hypersignal FLAIR et en hypersignal diffusion b1000 sans restriction sur la cartographie de l’ADC, sans prise de contraste après injection de Gadolinium. Figure (1). Un électroencéphalogramme a retrouvé un tracé d’organisation préservée, fait de rythme alpha postérieur moyennement volté, légèrement irrégulier, avec absence d’anomalie comitiale. Un dosage de la vitamine B1 érythrocytaire a été réalisé, en faveur d’une carence profonde en thiamine (taux de thiamine total à 79 mmol/l, pour un intervalle de normalité entre 126 et 250 mmol/l). La patiente a été supplémentée en thiamine à la posologie de 1 g/jour en intraveineux, pendant 10 jours, avec un relais par voie orale. Une IRM de contrôle faite après 3 mois a montré une nette régression de l’hypersignal bi thalamique ; une normalisation de la diffusion, une stabilité des hypersignaux FLAIR micronodulaires intéressant la substance blanche supra-tentorielle, aspécifique (Figure 2). La patiente s’est améliorée sur le plan neurologique dès les premières 48h. L’ataxie a complètement régressé dans les mois qui suivent, mais un nystagmus séquellaire persiste jusqu’à ce jour (trois ans après). REVUE DE LA LITTERATURE Les carences vitaminiques, essentiellement ceux du groupe B ; sont souvent responsables de troubles neurologiques et d’états confusionnels. Ces carences sont majoritairement associées à l’alcoolisme (1). Le déficit en thiamine peut s’observer chez des patients non alcooliques, dans la population pédiatrique et les adolescents, en cas de malnutrition, d’alimentation parentérale prolongée, de maladie gastro-intestinale, de diarrhée prolongée, vomissements récurrents, chirurgie bariatrique, hépatopathie, pathologie maligne, vomissements gravidiques et hémodialyse (19,13,18). Sur le plan anatomopathologique, l’EW est caractérisée par des suffusions hémorragiques associées à une prolifération gliale et à une démyélinisation au niveau des structures entourant le troisième ventricule, les corps mamillaires et les noyaux oculomoteurs. Des études de prévalence basées sur des autopsies montrent que l’EW est une maladie fréquente qui n’est souvent décelée qu’après le décès. A l’autopsie, on trouve des lésions caractéristiques chez environ 1,5% de la population générale et la prévalence augmente à 12,5% chez les patients alcoolo-dépendants (4,10,11) alors que 5 à 14% seulement des personnes atteintes sont identifiées avant leur décès (24). Les difficultés diagnostiques sont dues au fait que seulement 10% des patients atteints présentent simultanément la triade classique associant confusion, ataxie et ophtalmoplégie (21). D’autres signes cliniques comme des troubles cognitifs, une somnolence, un syndrome de Korsakoff (syndrome amnésique lié à des signes frontaux), un état stuporeux voire un coma, qui apparaissent chez 80% des patients présentant une EW, peuvent également être attribués à une intoxication alcoolique aiguë, à un syndrome de sevrage ou à un problème médical autre. L’ataxie est présente chez seulement 23% des patients et l’ophtalmoplégie chez 29%, d’autres symptômes tels qu’une hypotension ou hypothermie inexpliquée peuvent être présents (17,26). L’EW se manifeste donc souvent par un état confusionnel aspécifique entraînant chez 90% des patients le risque que le diagnostic soit manqué (21). Selon les recommandations de l’EFNS (European Federation of Neurological Societies), un dosage de thiamine doit être réalisé dès la suspicion clinique de l’EW, et une IRM doit être faite pour conforter le diagnostic (5). L’IRM montre une hyperdensité symétrique au niveau des corps mamillaires, du thalamus, de la matière grise périaqueducale, et des colliculi supérieur et inférieur (structure sous-corticale bilatérale située sur le toit du mésencéphale), cette hyperdensité est observée en T2 et en FLAIR (Fluid-Attenuated Inversion Recovery) (1). Le pronostic des patients insuffisamment ou non traités est sombre. Victor et al. ont observé une récupération complète chez seulement 16% des patients, avec une EW traitée avec des faibles doses parentérales de 50 à 100 mg de thiamine par jour, le taux de mortalité étant de 17 à 20% (26). Pour améliorer le pronostic des patients chez qui on diagnostique ou suspecte une EW, la thiamine doit être administrée aussitôt que possible à un dosage adéquat, et tout retard de la prise en charge augmente le taux de mortalité (3). DISCUSSION La thiamine est une vitamine exogène hydrosoluble, qui peut être stockée par le corps. C’est un cofacteur des enzymes de la voix d’oxydation des pyruvates. La carence en cette vitamine se manifeste cliniquement par une encéphalopathie de Wernicke, le diagnostic est clinique, ainsi un dosage normal en thiamine ne devrait pas retarder la supplémentation vitaminique immédiate. La triade caractéristique de l’EW était absente chez notre patiente comme dans 80% à 90% des cas décrits dans la littérature (12,15). Les troubles oculaires étaient au premier plan, avec la baisse de l’acuité visuelle, et une dysfonction oculomotrice, habituellement retrouvés chez 29% des patients atteints d’encéphalopathie de Wernicke (20,8). Les patients sous nutrition parentérale prolongée, nécessitent un apport plus important en thiamine afin de métaboliser leurs hydrates de carbone (2). Toutes les publications, mise à part une seule, ont conclu à ce que l’alimentation parentérale prolongée soit un facteur de risque primaire de l’encéphalopathie de Wernicke dans le contexte d’allogreffe (3). Notre patiente a reçu 6 semaines de nutrition parentérale exclusive en post allo-greffe, sans supplémentation en thiamine, avec apparition des symptômes à J42. Plusieurs auteurs préconisent une supplémentation en thiamine systématique en prophylaxie de l’EW, quoiqu’une publication d’un groupe brésilien, a mis en évidence le décès de 8 patients atteints d’EW, malgré une prophylaxie préalable par la thiamine à raison de 50mg par jour (3). Jusqu’à présent il n’y a toujours pas de consensus pour traiter l’EW, des essais randomisés et contrôlés doivent être menés afin de trouver la posologie et la durée optimale du traitement par thiamine, autant en curatif qu’en préventif. L’EFNS recommande 200 mg par voie intraveineuse 3 fois par jour, jusqu’à ce qu’il y ait stabilité des signes cliniques. Elle préconise également un suivi du dosage de la thiamine pendant au moins 6 mois (6). D’autre part, le NICE (National Institue for Health and Care Excellence) recommande une posologie de 500 mg ou 750 mg, 3 fois par jour pendant 5 jours. Une supplémentation par voie orale est recommandée après l’administration intraveineuse (6). Une amélioration clinique a été rapidement notée chez notre patiente dès l’initiation de la supplémentation en thiamine. L’ophtalmoplégie s’améliore en général dans les heures qui suivent la supplémentation, l’ataxie se résout en quelques jours, et la confusion mentale prend environ 3 à 4 semaines avant de s’arranger (9). Notre patiente a gardé un nystagmus séquellaire, seulement 20% des patients récupèrent complètement (3). Ce cas clinique met le point sur le rôle vital de la supplémentation en thiamine, chez les patients recevant une alimentation parentérale prolongée, surtout dans un contexte de néoplasie et de greffe. Les patients ayant subi une allogreffe sont à haut risque de développer une encéphalopathie de Wernicke, à cause de la malnutrition chronique, des nausées/vomissements chimio-induits, et de la forte consommation de thiamine par les cellules tumorales en croissance (16). CONCLUSION En conclusion, devant une symptomatologie neurologique et ophtalmologique, dans un contexte de dénutrition, de chimiothérapie lourde, de nutrition parentérale prolongée, de vomissements incoercibles, de chirurgie bariatrique, de néphropathie ou d’hépatopathie, une IRM cérébrale associée à un dosage de la vitamine B1, doivent être réalisés immédiatement. La supplémentation en thiamine devrait être débutée aussitôt que le diagnostic d’encéphalopathie de Wernicke est suspecté afin de prévenir la mortalité et la morbidité.
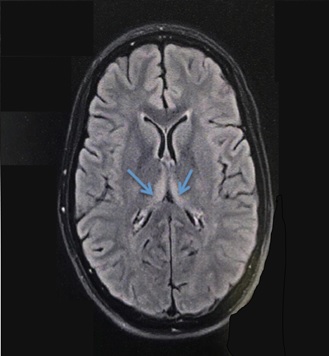 Figure 1 : IRM cérébrale montrant une lésion bithalamique ventrale et postérieure en hypersignal FLAIR. Cet aspect est évocateur d’une encéphalopathie de Wernicke
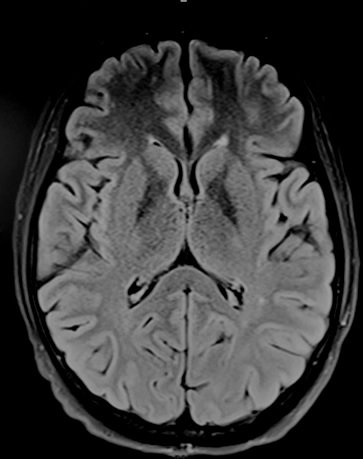 Figure 2 : IRM cérébrale, après 3 mois de supplémentation vitaminique, montrant une nette diminution de l’hypersignal bithalamique. RÉFÉRENCES
|
© 2002-2018 African Journal of Neurological Sciences.
All rights reserved. Terms of use.
Tous droits réservés. Termes d'Utilisation.
ISSN: 1992-2647