
|
|
|
CLINICAL STUDIES / ETUDES CLINIQUES
ENCEPHALOPATHIE EPILEPTIQUE INFANTILE PRECOCE D’ETIOLOGIE ANOXO-ISCHEMIQUE AU CHU CAMPUS DE LOME (TOGO)
EARLY INFANTILE EPILEPTIC ENCEPHALOPATHY WITH ANOXO-ISCHEMIC ETIOLOGY AT UNIVERSITY HOSPITAL CAMPUS OF LOME (TOGO)
E-Mail Contact - DIATEWA Josué Euberma :
lejd01@gmail.com
RÉSUMÉ L’Encéphalopathie Epileptique Infantile Précoce (EEIP) ou syndrome d’Ohtahara, qui est une forme rare d’encéphalopathie néonatale, présente des caractéristiques électro-cliniques, thérapeutiques et évolutives particulières. Ses étiologies, essentiellement structurelles, sont plus malformatives qu’anoxo-ischémiques pour la majorité des cas décrits dans la littérature. Nous rapportons les deux premiers cas d’EEIP d’étiologie anoxo-ischémique, confirmés par la tomodensitométrie cérébrale et suivis dans le service de neurologie depuis l’âge de 2 et 5 mois, respectivement. Les patients avaient des antécédents de souffrance cérébrale néonatale et, jusqu’à l’âge d’un an, présentaient une évolution clinique sévère marquée par la persistance de tornades de spasmes toniques en extension (plus de 20 crises par jour) et un retard de développement psychomoteur, malgré la prise de phénobarbital, de carbamazépine ou de valproate de sodium et de méthylprednisone. A l’âge de huit mois et un an, l’électroencéphalogramme de sommeil était toujours de type suppression-burst, tandis que celui de veille se transformait en hypsarythmie (syndrome de West). Mots-clés : Syndrome d’0htahara ; Encéphalopathie épileptique infantile précoce ; Syndromes épileptiques ; Anoxie cérébrale ; Lomé. ABSTRACT Early Infantile Epileptic Encephalopathy (EIEE) or Ohtahara syndrome is a rare form of neonatal encephalopathy. It presents particular electro-clinical, therapeutic and outcome features. Its etiologies are essentially structural and more malformative than anoxo-ischemic for most of the cases which are described in literature. We report the first two cases of EIEE with anoxo-ischemic etiology, which were confirmed by CT-scan and followed up in the department of neurology since 2 and 5 months old, respectively. Patients had a neonatal cerebral pain history and, up to one year old, presented a severe outcome which was characterized by the persistence of tonic spasms tornadoes in extension (more than 20 seizures per day) and psychomotor development delay, despite taking phenobarbital, carbamazepine or sodium valproate and methylprednisone. At eight months and one year old, the electroencephalogram of sleep presented still suppression-burst pattern, while that of awakening was transformed into hypsarrhythmia (West’s syndrome). Key words: Ohtahara Syndrome; Early Infantile Epileptic Encephalopathy; Epileptic Syndromes; Cerebral Anoxia; Lomé. INTRODUCTION L’Encéphalopathie Epileptique Infantile Précoce (EEIP) ou syndrome d’Ohtahara est un syndrome épileptique particulier en raison de la survenue précoce des crises associées (période néonatale), l’évolution électro-clinique atypique, le pronostic sévère et l’étiologie multifactorielle [6]. A ce jour, seuls des cas isolés sont rapportés dans la littérature [6]. Pour ces derniers, l’étiologie malformative est rapportée dans plus de 50% des cas, comparativement à celle résultant d’une anoxie cérébrale (18% des cas) [3,6,8]. A cet effet, nous rapportons les deux premiers cas documentés de syndrome d’Ohtahara post anoxo-ischémique, confirmés par la tomodensitométrie cérébrale au CHU Campus de Lomé (Togo). OBSERVATIONS Observation 1 Le nourrisson K.L., de sexe féminin, était suivi dans le service de neurologie du CHU campus de Lomé depuis l’âge de cinq mois et jusqu’à l’âge d’un an pour des spasmes toniques en extension prédominants à gauche (Figure 1). Ces crises évoluaient depuis le 12e jour de vie, sans fièvre. La fréquence des crises étaient supérieure à vingt par jour. Aucun facteur déclenchant n’avait été retrouvé. La patiente avait bénéficié d’un traitement antérieur traditionnel (breuvage) et d’un médicament antiépileptique (phénobarbital à la dose de 5 g/kg/j) administré deux mois auparavant par un pédiatre. La patiente était la dernière d’une fratrie de quatre enfants, tous en bonne santé apparente. Aucune affection familiale congénitale n’avait été rapportée. Elle était née par césarienne pour souffrance fœtale aiguë avec un score d’APGAR à 6-7-9. A son admission, l’examen clinique objectivait une bonne croissance staturo-pondérale (poids: 6500 g ; taille: 64 cm; périmètre crânien : 43 cm) et une apyrexie (température: 37° C). Aucun signe dysmorphique, ni d’anomalies cutanées, n’étaient retrouvés. Au plan neurologique, on notait une hypotonie axiale. La patiente était incapable de passer du décubitus dorsal au décubitus ventral. La préhension volontaire était faible. La manifestation des émotions (rire et petits cris intentionnels de joie) était peu marquée, ainsi que la recherche du regard à l’appel. L’EEG de veille et de sommeil montrait un pattern de suppression-burst avec des bouffées d’ondes lentes, mêlées de pointes et pointes-ondes, plus amples à droite. Les périodes de bouffées étaient plus longues que les périodes suppressives (Figure 2). Le scanner cérébral révélait des lésions d’anoxie pariéto-occipitales bilatérales (Figures 3 a et b). Le bilan métabolique était normal. Les médicaments administrés étaient la carbamazépine (10 mg/kg/j) et le phénobarbital (5 mg/kg/j). A l’âge de huit mois, l’évolution était marquée par la persistance de la fréquence élevée des crises et par le retard psychomoteur plus prononcé (position assise impossible, impossibilité de saisie franche d’un jouet, pince pouce-index difficile, cris aigus). L’EEG de sommeil était superposable au précédent, tandis que celui de veille se transformait en hypsarythmie (syndrome de West) (Figure 4). Le méthylprednisone (1-3 mg/kg/j) avait été administré durant deux mois, avec dose régressive. La kinésithérapie motrice avait été réalisée. A un an, une régression de la fréquence des crises avait été notée (dix par jour), avec amélioration minime du développement psychomoteur caractérisée par la direction de la main vers un objet et la saisie, des cris pour attirer l’attention, une expression meilleure des émotions. Observation 2 Le nourrisson K.A., de sexe féminin, était suivi dans le service de neurologie du CHU campus de Lomé depuis l’âge de deux mois et jusqu’à un an pour des spasmes toniques en extension, asymétriques et prédominants à gauche. Ces crises évoluaient depuis le 10e jour de vie, sans fièvre. La fréquence des crises était supérieure à quinze par jour. Aucun facteur déclenchant n’avait été retrouvé. La patiente avait bénéficié d’un traitement antérieur à base de phénobarbital (5 mg/kg/j), prescrit par un pédiatre. La patiente était la dernière d’une fratrie de deux enfants en bonne santé apparente. Elle était née par voie basse avec forceps, malgré le dépassement de terme (41 semaines d’aménorrhée). Le score d’APGAR était de 4-7-9. L’examen clinique à trois mois objectivait une bonne croissance staturo-pondérale (poids: 5350 g ; taille: 59 cm; périmètre crânien : 39 cm) et une apyrexie (température: 37° C). Aucun signe dysmorphique, ni d’anomalies cutanées, n’avaient été retrouvés. Au plan neurologique, on notait une hypotonie axiale, une absence de jeux avec les mains en les joignant, un langage limité aux gazouillis, une absence de poursuite oculaire des objets en mouvement ou en réponse à des sons. L’EEG de veille et de sommeil montrait un pattern de suppression-burst similaire à celui de la figure 2. Le scanner cérébral révélait des lésions d’anoxie fronto-pariétales bilatérales (Figures 3 c et d). Le bilan métabolique était normal. Le valproate de sodium (30 mg/kg/j) et le phénobarbital (5 mg/kg/j) avaient été administrés à la patiente. A l’âge de six mois, la fréquence des crises était toujours élevée et le retard psychomoteur plus prononcé (position assise impossible, de même que la saisie franche des objets et l’imitation des sons; humeurs peu expressives). L’EEG de veille et de sommeil était similaire au précédent. Le méthylprednisone (1-3 mg/kg/j) avait été administré durant deux mois, avec dose régressive. La kinésithérapie motrice avait été recommandée, mais non réalisée. A huit mois, une régression de la fréquence des crises avait été notée (cinq par jour), sans amélioration du développement psychomoteur. L’EEG de veille était de type hypsarythmie (Syndrome de West) (Figure 4). DISCUSSION L’EEIP est peu rapportée en Afrique subsaharienne en raison vraisemblablement de son évolution péjorative [1] et de la stigmatisation liée à l’épilepsie [2,4]. La rareté de l’étiologie anoxo-ischémique de ce syndrome pourrait se justifier entre autres par l’imagerie médicale (TDM et IRM) non disponible et/ou d’accès difficile. Les caractéristiques résultant des données du présent travail sont principalement l’évolution électro-clinique sévère et les particularités liées aux critères diagnostiques. Les EEIP post anoxo-ischémiques présentent une grande variété sémiologique des crises. Les spasmes n’y sont pas rapportés [9]. Dans la présente étude, les spasmes observés constituent une situation exceptionnelle. Le mécanisme physiopathologique de ces spasmes n’est pas encore bien élucidé. Par analogie faite aux spasmes dans le syndrome de West [5], leur survenue dépend plutôt de l’âge et de la localisation anatomique des lésions cérébrales que de l’étiologie. De cet fait, il y a une forte probabilité qu’ils apparaissent dans les cas de lésions corticales postérieures vers trois ou quatre mois d’âge et de lésions frontales vers six mois d’âge. Durant cette période critique, le développement cérébral est maximal [5]. Ces observations confirment les données cliniques et radiologiques de nos nourrissons. Les lésions scanographiques, rapportées dans cette étude, évoquent prioritairement une anoxo-ischémie. Une séquelle infectieuse ou une anomalie de migration neuronale ne peut pas être éliminée de façon formelle. En effet, l’IRM cérébrale, examen morphologique de choix dans l’exploration des épilepsies chez l’enfant, n’a pas été réalisée en raison des ressources financières limitées des parents. Un contrôle efficace de la fréquence des crises au cours du syndrome d’Ohtahara s’effectue après utilisation du valproate de sodium en première intention, de barbiturique en seconde intention et de corticostéroïdes en cas d’inefficacité des médicaments antiépileptiques (MAE) [8]. Malheureusement, les ressources financières réduites des parents ont limité la mise en œuvre de cette stratégie thérapeutique chez nos nourrissons. D’où, l’administration d’une bithérapie en première intention (phénobarbital associé à la carbamazépine ou au valproate de sodium). Les indications thérapeutiques de la carbamazépine sont principalement représentées par les crises focales avec ou sans bilatéralisation secondaire [10]. Aucune contre-indication de la carbamazépine dans le traitement des spasmes n’est rapportée dans la littérature. Cependant, un risque d’aggravation des spasmes a été rapporté, imposant ainsi des précautions d’emploi [10]. Dans notre étude, les spasmes asymétriques observés ont été assimilés aux crises généralisées à début focal. La catégorisation de ces spasmes a été motivée par la nécessité d’initier un traitement efficace en raison de l’insuffisance de ressources financières des parents, laquelle a fortement limité l’utilisation précoce du valproate de sodium ou du méthylprednisone, conformément aux recommandations de la littérature [8]. L’administration de la méthylprednisone durant deux mois, à dose régressive, conformément aux recommandations de la littérature [8], a relativement amélioré l’évolution clinique de nos nourrissons. Les lésions cérébrales de ces derniers étant étendues, nous ne pouvions pas obtenir un véritable contrôle des crises ; le risque d’invalidité est sévère. Des auteurs ont rapporté la disparition totale des suppressions-burst avant l’âge de six mois [6] et une transformation en Syndrome de West (73% des cas) après trois mois [8]. La persistance des suppressions-burst et la transformation en syndrome de West entre huit mois et un an d’âge chez nos nourrissons, suggèrent que l’origine et l’étendue des lésions cérébrales au cours du syndrome d’Ohtahara ont un impact sur l’évolution électro-clinique de ce dernier. Par ailleurs, l’induction iatrogène et tardive du syndrome de West ne peut pas être exclue [10], en particulier chez le nourrisson KL. En effet, le risque d’induction du syndrome de West par la thérapie à la carbamazépine ou apparentés chez des patients souffrant de crises focales a été rapporté par divers auteurs [7,10]. Malheureusement, nous n’avions pas suspendu la prise de carbamazépine, ce qui aurait pu mettre en évidence le caractère inducteur du médicament chez le nourrisson KL. Toutefois, l’amélioration de la fréquence des crises chez les deux patientes sous méthylprednisone et la présence de nombreux facteurs de mauvais pronostic (début tardif de la maladie, durée prolongée des spasmes, absence d’amélioration du développement psychomoteur, retard de corticothérapie, lésions cérébrales étendues) suggèrent que le caractère inducteur de la carbamazépine chez le nourrisson KL est probablement minime. De plus, la prise de carbamazépine à faible dose (10 mg/kg/jour) aurait probablement peu d’effet inducteur. CONCLUSION L’EEIP d’étiologie anoxo-ischémique est une pathologie rare et caractérisée par un pronostic sévère. Cette étiologie, en rapport avec la souffrance cérébrale néonatale, reflète la persistance des difficultés de soins pré-, péri- et post-natals rencontrée en Afrique subsaharienne. L’exploration neuroradiologique par l’IRM est nécessaire et indispensable pour un meilleur diagnostic étiologique. La sensibilisation et la recherche scientifique au moyen d’études multicentriques restent d’actualité afin de mieux individualiser les cas d’EEIP et en assurer une prise en charge adéquate. DECLARATION D’INTERET Les auteurs déclarent n’avoir aucun conflit d’intérêt en rapport avec cet article.
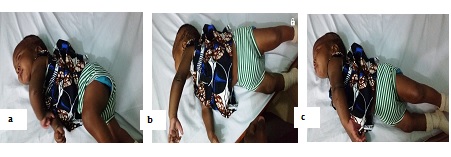 Figure 1 : Séquence montrant les spasmes en extension asymétrique
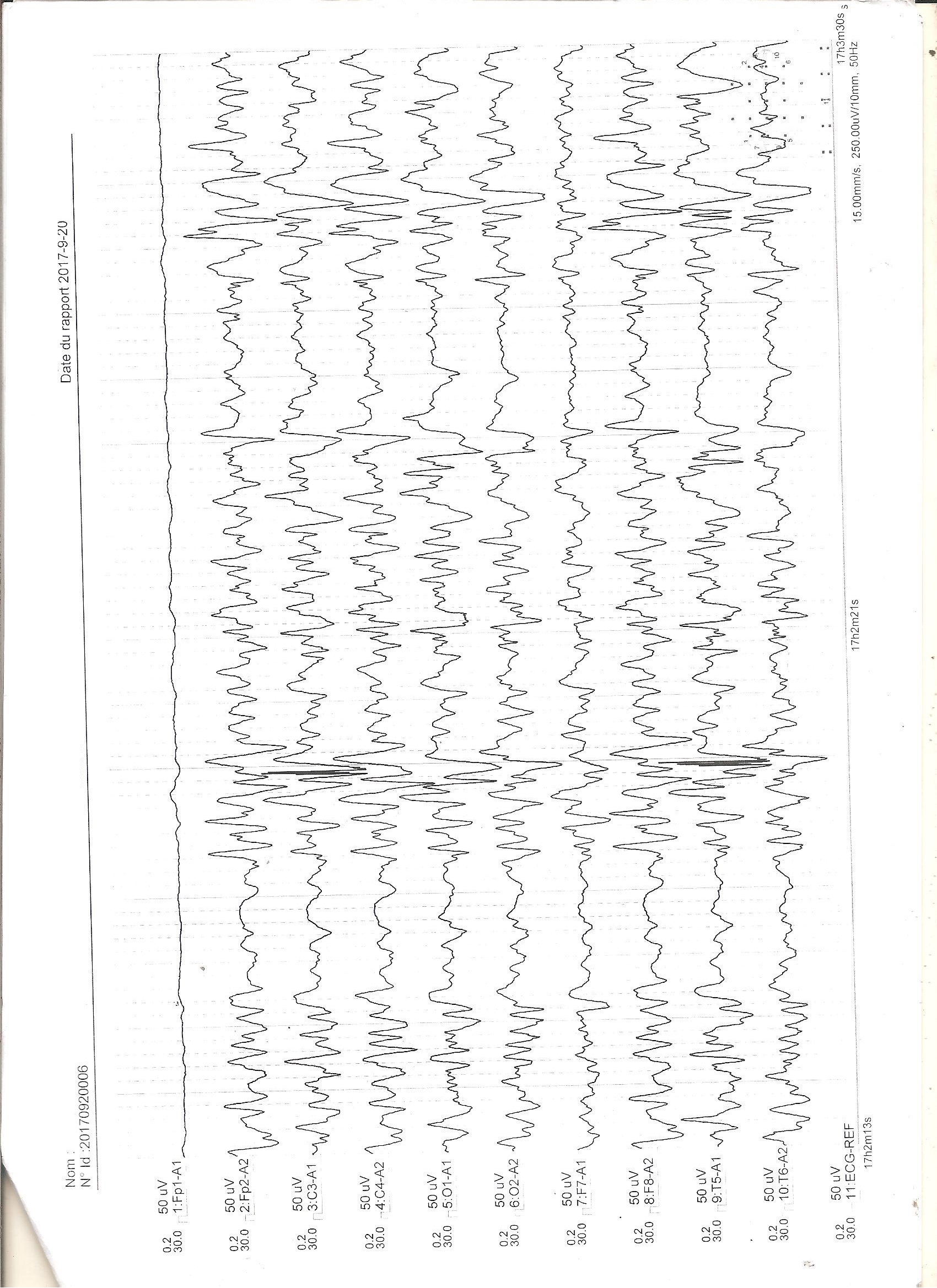 Figure 2 : EEG de veille et de sommeil de type suppression-burst
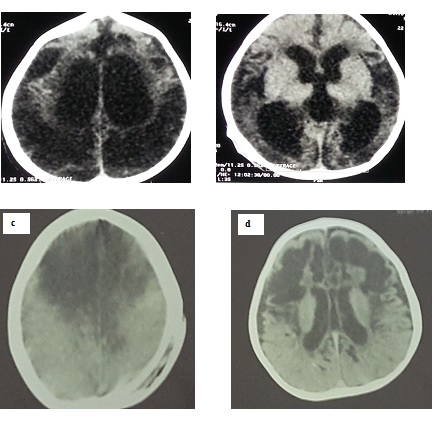 Figure 3: Scanner cérébral sans injection de produit de contraste, en coupes axiales : lésions d’anoxie cérébrale, pariéto-occipitales bilatérales (a et b) et fronto-pariétales bilatérales (c et d), respectivement, chez les nourrissons 1 et 2
 Figure 4 : Hypsarythmie atypique chez les nourrissons 1 et 2
REFERENCES
|
© 2002-2018 African Journal of Neurological Sciences.
All rights reserved. Terms of use.
Tous droits réservés. Termes d'Utilisation.
ISSN: 1992-2647