
|
|||||||||||||||||||||
|
CLINICAL STUDIES / ETUDES CLINIQUES
EPILEPSIE-ABSENCES DE L’ENFANT ET DE L’ADOLESCENT AU SENEGAL ASPECTS EPIDEMIOLOGIQUES, DIAGNOSTIQUES, THERAPEUTIQUES ET PRONOSTIQUES
ABSENCES EPILEPSY OF CHILDREN AND TEENAGER IN SENEGAL EPIDEMIOLOGICAL, DIAGNOSTIC, THERAPEUTIC AND PROGNOSTIC ASPECTS
E-Mail Contact - FALL Maouly :
fall.maouly@gmail.com
RESUME Description: L’épilepsie-absence est un syndrome épileptique généralisé, fréquent, de cause présumée génétique, caractérisé par la survenue d’épisodes fréquents de rupture brutale et complète du contact sans perte de tonus. Objectif: L’objectif était de décrire l’épidémiologie et les caractéristiques cliniques de l’épilepsie-absence chez l’enfant et l’adolescent. Méthode: Nous avons mené une étude transversale longitudinale, portant sur 53 patients, de décembre 2003 à mars 2014 en colligeant les données épidémiologiques, diagnostiques, thérapeutiques et évolutives. L’analyse des données s’est faite avec le logiciel statistique CSPro 5.0 avec calcul de fréquences et moyennes. Résultats: Cinquante trois patients dont 42 enfants et 11 adolescents ont été colligé avec un âge moyen de 10 ans. L’âge moyen de début des crises était de 6,8 ans chez les enfants et 12,4 ans chez les adolescents. On notait une prédominance féminine à 52,4 % chez les enfants et masculine à 63,6 % chez les adolescents. Une consanguinité parentale était retrouvée dans 55,5 % chez l’enfant et 37,5 % chez l’adolescent. Quarante pourcent des enfants présentaient des antécédents familiaux d’épilepsie. Les crises survenaient spontanément dans 86,8 % des cas avec une durée moyenne de 10 secondes environ. Le Valproate de sodium a été utilisé chez tous nos patients avec une maitrise dans 81,6 % à trois mois. L’évolution de l’épilepsie-absence était globalement satisfaisante avec des difficultés d’apprentissage observées chez 22,6 % des patients. Conclusion: L’épilepsie-absence est fréquente, de causes multifactorielles et peut retentir sur le devenir psychosocial. Mots clés : adolescent, enfant, épilepsie-absence, prise en charge, Sénégal ABSTRACT Background: Absence-epilepsy is a generalized epileptic syndrome, frequent, of presumed genetic cause, characterized by the occurrence of frequent episodes of sudden, profound impairment of consciousness without loss of body tone. Objective: The objective was to describe the epidemiology and clinical features of absence-epilepsy in children and adolescents. Methods: We conducted a longitudinal cross-sectional study of 53 patients from December 2003 to March 2014, collecting epidemiological, diagnostic, therapeutic and evolutionary data. Analysis of data was done with CSPro 5.0 statistical software with frequency and averaging calculations. Results: Fifty-three patients including 42 children and 11 adolescents were enrolled with an average age of 10 years. The mean age of onset of seizures was 6.8 years in children and 12.4 years in adolescents. We found female prevalence of 52.4% in children and male prevalence of 63.6% in teenagers. Parental consanguinity was found in 55.5% in children and 37.5% in adolescents. Forty percent of children had a family history of epilepsy. Seizures occurred spontaneously in 86.8% of cases with an average duration of about 10 seconds. Sodium valproate was used in all our patients with a mastery in 81.6% at three months. The evolution of absence-epilepsy was generally satisfactory but learning difficulties were observed in 22.6% of patients. Conclusion: Absence-epilepsy is common, have multifactorial causes and can affect the cognitive outcome. Keywords: care, children, absence epilepsy, Senegal, teenager INTRODUCTION L’incidence des épilepsies de l’enfant de 4 à 16 ans est de 82 pour 100.000 habitants, avec une prévalence de 4,9 ‰ (11). Selon la LICE (2), le diagnostic de l’épilepsie-absence de l’enfant et de l’adolescent (EAEA) repose sur des critères bien définis qui font d’elle un syndrome distinct dont les composantes génétiques et développementales sont clairement établies (1,6,8,13,24). Les EGI dont les épilepsies-absences de l’enfant (EAE) ont souvent été présentées comme une entité d’épilepsie à évolution normale sous traitement et sans séquelles. Toutefois, des troubles neuropsychologiques sont parfois associés (5,9,10). En France, les épilepsie-absences de l’enfant (EAE) constituent 11% des épilepsies de l’enfant avec une nette prédominance féminine (60 à 70%) (19). A Marrakech, une étude réalisée en 2010 sur 592 enfants épileptiques avait permis de mettre en évidence 12% de cas d’EAE (4). Au Sénégal, en 2006, 16,34% d’épilepsies généralisées idiopathiques (EGI) avait été constaté sur un total de 459 patients. Dans cette série, les EAE représentaient le syndrome le plus fréquent avec 28% des EGI (versus 2,34% pour les EAA) (20). L’objectif général de cette étude était de décrire les aspects épidémiologique, diagnostique, thérapeutique et pronostique de l’épilepsie-absence de l’enfant et de l’adolescent (EAEA) à la Clinique Neurologique du Centre Hospitalier Universitaire National de Fann et à l’Hôpital d’Enfants Albert Royer à Dakar. METHODOLOGIE Nous avons mené une étude transversale longitudinale de patients reçus en consultation de décembre 2003 à mars 2014 à la Clinique Neurologique du Centre Hospitalier Universitaire National de Fann et à l’Hôpital d’Enfants Albert Royer à Dakar. Les objectifs spécifiques étaient d’étudier les aspects épidémiologiques des EAEA au Sénégal, d’en décrire les aspects électro-cliniques, d’apprécier l’efficacité et la tolérance des molécules utilisées dans le traitement de l’EAEA et d’étudier les aspects évolutifs de la maladie, essentiellement son retentissement sur la scolarité des patients. Nous avons sélectionné les patients diagnostiqués EAE (Epilepsie-absence de l’enfant) ou EAA (épilepsie-absence de l’adolescent) durant la période d’étude, suivant nos critères de définition : ◊ Crises d’absence typiques ◊ Début entre 4 à 10 ans pour l’EAE et à partir de 10 ans pour l’EAA ; ◊ Développement psychomoteur (DPM) normal ; ◊ Examen neurologique strictement normal ; ◊ Electroencéphalographie (EEG) typique d’une EA (bouffées de pointes ou de pointe-ondes d’environ 3 Hz à début et fin brusque). Les données ont été recueillies grâce à un questionnaire fait avec l’aide d’un neuro-épidémiologiste. Les paramètres étudiés étaient d’ordre sociodémographiques (sexe, âge), cliniques (âge de début et âge au moment du diagnostic, antécédents familiaux d’épilepsie et/ou de consanguinité parentale, les caractéristiques et signes associés des absences), thérapeutique (molécules utilisées et réponse au traitement) et pronostique avec un bilan neuropsychologique de l’enfant (évaluant le développement à la fois social, affectif et intellectuel de l’enfant, en recherchant des difficultés dans le milieu scolaire voire des échecs dans ce domaine, avant et après traitement) précédé et complété par un entretien avec les parents dans ce sens. L’analyse des données s’est faite avec le logiciel statistique CSPro 5.0 avec calcul de fréquences et moyennes. RESULTATS Nous avons colligé 53 patients dont 45 à la Clinique Neurologique du CHNU de Fann. L’EAE concernait 42 patients soit une proportion de 79,2%, et l’EAA 11 patients (soit 20,8%). L’âge moyen des patients au moment du diagnostic était de 10 ans pour l’ensemble des patients (avec des extrêmes de 5 ans et 19 ans), et de 9 ans versus 13,7 ans respectivement pour l’EAE et l’EAA. L’âge moyen de début des crises des cas d’EAE était de 6,8 ans, versus 12,4 ans pour l’EAA et 7,9 ans sur l’ensemble de la cohorte. Le sex-ratio était de 1 sur l’ensemble de la série. On notait respectivement une prédominance féminine à 52,4% pour les cas d’EAE et masculine à 63,6% sur les formes d’EAA (Figure1). Nos patients évoluaient dans des familles majoritairement monogames (64,2%) aux parents instruits dans 90,6% des cas. Une consanguinité parentale était retrouvée dans 55,5% des cas d’EAE (soit 15 patients) et 37,5% des cas d’EAA (soit 3 patients) et était globalement de 33,9%. Pour l’EAE, les antécédents familiaux d’épilepsie étaient retrouvés dans 40% des cas (soit 12 patients). Les antécédents personnels, ne concernaient que les patients avec EAE, et étaient caractérisés par 3 cas de CGTC et 1 cas de myoclonie. Les crises d’absence survenaient spontanément dans la majorité des cas: soit globalement dans 86,8% des cas. L’appréciation du nombre de crises d’absence par jour n’était que subjective, car les parents n’arrivaient pas à le dire avec exactitude. Les crises d’absence étaient estimées à maximum 10 par jour pour la majorité des cas (67,9%) correspondant respectivement à 1/3 des cas d’EAE et d’EAA. La durée des crises était estimée à environ 10 secondes généralement. La rupture de contact était complète dans tous les cas et isolée dans 83,3% des cas pour l’EAE et 72,7% dans l’EAA. Les crises d’absence étaient accompagnées d’automatismes dans 26,4% des cas, principalement au cours de l’EAA (5/11 patients EAA contre 9/42 EAE). Ces automatismes, essentiellement moteurs, étaient faits de léchage des lèvres, de mâchonnement et d’éléments végétatifs (émission d’urines et hypersalivation). D’autres types de crises étaient associés chez 8 patients : myoclonies et CGTC. Dans la série, ceux qui en présentaient, avaient l’une ou l’autre des crises. Ces crises étaient apparues dans le courant de la maladie épilepsie-absence (Tableau 1). Dans les EA, les caractéristiques de l’EEG étaient typiques chez tous les patients avec des bouffées de pointes-ondes (BPO) à 3 Hz hypersynchrones généralisées et paroxystiques sur un rythme de fond normal, de durée variable, déclenchées ou majorées par l’hyperventilation. Dans notre série, les bouffées duraient entre 4 et 10 secondes dans l’EAE et l’EAA respectivement chez 25 patients (59,5%) et 6 patients (54,6%). Les bouffées étaient majoritairement spontanées et/ou accentuées par l’hyperpnée dans les cas d’EAE. L’imagerie cérébrale n’était pas systématique chez nos patients. Toutefois, elle fut faite et revenue normale chez 7 patients [1 IRM cérébrale pour 1 EAE et de 6 tomodensitométries (TDM) cérébrales dont 1 cas d’EAA] qui présentaient en plus d’autres types de crises tels des myoclonies ou CGTC, ou une réponse retardée au traitement. Tous les patients étaient systématiquement traités en monothérapie par valproate de sodium (VPA) dès le diagnostic posé ou supposé, avec une régression complète des crises d’absence constatée chez 48 patients (90,6%). Les 5 patients, à évolution défavorable sous VPA, ont été stabilisés après adjonction d’Ethosuximide (ESM). Parmi eux, 3 appartenaient à la même famille, avec une notion de consanguinité parentale et d’épilepsie familiale. Sur les 53 patients sous VPA, seuls 3 cas d’EAE (5,7 %) avaient présenté une obésité; d’autres effets secondaires n’ont pas été rapportés. Au terme des 3 premiers mois de traitement, 38 patients (71,7%) ne faisaient plus de crises d’absence : 31 cas d’EAE et 7 cas d’EAA. Les délais allant jusqu’à 1 an concernaient les patients qui avaient eu une réponse retardée au traitement et chez qui des associations de molécules avaient été nécessaires (figure 2). Ces délais ont été proposés aux parents des patients par souci d’harmonisation. Dans la réalité, certains patients, surtout ceux présentant l’EAE avait vu leurs crises s’arrêter en un mois. Le contrôle de l’EEG était normal chez 42 patients et étaient revenu anormal chez 7 patients (6 EAE et 1 EAA) dont les crises n’avaient pas été maitrisées au moment du contrôle EEG ou qui présentaient d’autres types de crises. Ces tracés EEG étaient caractérisés par une persistance des paroxysmes de pointes-ondes, non plus sous forme de longues décharges, mais de courtes bouffées, obtenues parfois à la deuxième ou troisième HPN. Les patients inscrits dans les écoles française et coranique étaient respectivement 47 (88,6%) et 3 (5,7%). Seuls 3 patients n’avaient pas été scolarisés dans notre série. Des difficultés d’apprentissage, soit concomitantes des absences ou apparaissant au cours de l’évolution de la maladie, ont été observées chez 22,6% de l’ensemble des patients (soit 12 patients dont 2 cas d’EAA), et 23,4% des patients (11) inscrits à l’école française (figure 3). Ces difficultés d’apprentissage n’ont toutefois pas été objectivées par des tests neuropsychologiques. Chez les patients avec difficultés d’apprentissage, 5 (45,4%) avaient une scolarité retardée et 3 (27,3%) étaient exclus. Parmi les patients sans difficultés d’apprentissage, 7 (19,4%) avaient un retard de scolarité et 2 (5,6%) exclus (Figure 3). DISCUSSION La fréquence des EAE est plus élevée que celle des EAA (16). Notre étude le conforte avec une fréquence des EAE évaluée à 79,2% des cas versus 20,8% pour les EAA. L’âge moyen du début des crises pour les EAE était de 6,8 ans et 12,4 ans pour les EAA dans notre série ; ce qui reste conforme aux données de la littérature, respectivement entre 6-7 ans et 10-12 ans (15, 20, 21). La prédominance féminine constatée dans notre série pour l’EAE (52,4%) était retrouvée dans la plupart des études (8, 15, 16, 21). Mais pour l’EAA, c’était plutôt le genre masculin qui prédominait avec un pourcentage de 63,6%. Dans la littérature, la distribution des deux sexes est classiquement équivalente pour l’EAA. Cependant, certaines études montrent une prédominance masculine de l’ordre de 55%, et d’autres, celle féminine à 53% (15). La scolarisation des patients a connu un taux assez élevé dans notre étude, 94,3% avec 88,6% dans les écoles françaises et 5,7% dans les écoles coraniques. L’EA n’a donc pas été un facteur limitant d’inscription des patients à l’école. Ndiaye et al. avaient retrouvé la même fréquence dans sa série concernant les EAE, 95,2% avec 85,7% dans les écoles françaises et 9,5% dans les écoles coraniques (20). Le niveau d’instruction des parents était assez remarquable dans notre étude (90,6%). Elle nous semble contributive d’une évolution favorable pour une pathologie qui a besoin d’être comprise dans ses manifestations, sa prise en charge et son suivi. La base génétique de l’EA a été confirmée dans la littérature (18,19). La consanguinité majore le risque de survenue d’épilepsies idiopathiques et ce risque est multiplié par trois si le parent du premier degré est épileptique (20). Dans notre étude, elle était retrouvée dans 55,5% des cas d’EAE, versus 37,5% des cas d’EAA; et était de 33,9% pour l’ensemble de la série. Dans la littérature, des études établissant l’influence de l’endogamie dans cette pathologie ne sont pas courantes. Cependant, Ndiaye M., dans le groupe des épilepsies idiopathiques avait retrouvé 23,7% de consanguinité parentale (20). Notre taux est également supérieur aux caractéristiques démographiques du Sénégal qui compte 41% de mariages endogamiques. Notre taux plus élevé pourrait s’expliquer par l’homogénéité de notre série avec exclusivement des EA, qui sont supposées être d’hérédité mendélienne à transmission dominante (22,23). La présence de consanguinité parentale justifie l’existence d’antécédents familiaux d’épilepsies chez les patients souffrant d’EA. Nous les avons retrouvés dans 40% pour l’EAE et 37,5% pour l’EAA. Plusieurs études ont rapporté des antécédents familiaux dans les EA. Urena-Hornos T et al. avaient constaté dans une même famille, 9 cas d’EA avec âge de début variable (23). Notre proportion d’antécédents familiaux pour l’EAE est nettement supérieure à celui d’Ould C.H. (35,7%) (21), du fait probablement du nombre plus important d’EAE de notre série augmentant ainsi les possibilités d’enregistrement de cas familiaux d’épilepsies. Les crises d’absence, typiques chez tous nos patients étaient parfois accompagnées d’automatismes dans 26,4% des cas. Des taux plus élevés d’automatismes associés sont rapportés dans la littérature (21,23). Mais ce sont des manifestations diversement appréciées par les parents et même le personnel de santé, souvent en raison de leur discrétion et surtout parce qu’elles ne sont souvent pas présentes à toutes les crises. Les crises d’absence survenaient le plus souvent spontanément mais aussi lors d’émotions à type de contrariété ou tristesse respectivement chez 9,5% des EAE et 27,3% des EAA. Le taux plus élevé chez les patients présentant l’EAA pourrait s’expliquer par leur âge péri-pubertaire avec tout le remodelage psychologique qui caractérise cette période. D’autres facteurs comme un manque de sommeil fréquents à cette période du fait des abus de jeux-vidéos et la fatigue sont rapportés dans la littérature (17,18,19,21,23). Les crises étaient moins fréquentes chez la majorité de nos patients (5-10 crises par jour) comparé à la littérature (21,23). Ceci reflète une certaine subjectivité dans l’appréciation de la fréquence des crises par les parents, qui ont déjà du mal à accepter ces épisodes comme de véritables crises épileptiques (distraction, simulation, inattention, exprès, …). Les autres types de crises retrouvées dans notre étude étaient essentiellement des myoclonies et CGTC. Dans les EAA, on n’avait aucun cas de myoclonie, juste des CGTC (36,4%). Ceci avait l’avantage de faciliter notre classification et nous permettait d’exclure systématiquement toute possibilité d’intrusion de cas d’épilepsie myoclonique juvénile dans notre cohorte, car dans cette entité d’EGI, les crises d’absence pouvaient aussi être associées aux myoclonies dans le tiers des cas (19). Cependant dans la littérature, certains cas d’EA et particulièrement d’EAA sont associés aux myoclonies. Ces myoclonies, comme les CGTC surviennent souvent dans le cours de la maladie et sont des facteurs pronostiques car imposant des bithérapies avec leurs difficultés d’interactions thérapeutiques (3,7,14). Les CGTC retrouvés aussi bien dans l’EAE que dans l’EAA sont aussi rapportées par plusieurs études. Ould C. H. (21) en a retrouvé dans une proportion de 35,71% pour les EAE avec une prédominance masculine, versus 36,4% dans notre cohorte. Ainsi, dans la littérature le genre masculin est décrit comme facteur de risque de l’association absence et CGTC (3,23). Dans notre série, la molécule de 1ère intention était le valproate de sodium (VPA), prescrit systématiquement en monothérapie chez tous nos patients. Il avait aidé à maîtriser les crises dans une proportion de 84,9%, versus une moyenne de 75% des cas pour Hacen (21). Le recours élevé au VPA s’expliquait par sa grande accessibilité au Sénégal mais surtout son action bénéfique sur les autres types de crises parfois associées (20) et sa tolérance (3 cas d’obésité rapportés). Les indications thérapeutiques dans l’EA ont été bien codifiées par un essai américain publié en 2010 (1) qui mettait en avant le VPA et l’ESM au même niveau avec un taux de disparition des absences, quasi-égal, respectivement 58% et 53% des cas, alors que la LTG n’en avait que 29%. Donc, le véritable choix doit être guidé par la tolérance des molécules et il n’y avait pas de différence sur les effets secondaires entre les groupes étudiés pour les trois molécules. Mais les patients sous VPA avaient présenté plus de troubles attentionnels (49%) que ceux sous ESM (33%). Aussi, on recommande l’ESM en première intention dans les EAE, tandis que dans l’EAA on privilégie le VPA et la LTG car l’ESM a un effet limité sur les CGTC (23). Les crises se sont arrêtées sous traitement dans les trois premiers mois chez la majorité des patients dans l’EAE (81,6%) et dans l’EAA (63,6%). Ould C. H. avait rapporté une totale maîtrise des absences dans 89,2% des cas, sans préciser le délai d’arrêt des crises (21). Toutefois, la rapide maîtrise des crises n’exclut pas l’accompagnement et le suivi des patients pour dépister des crises motrices éventuelles ou la survenue de troubles neuropsychologiques. Le protocole de suivi des EA impose que des EEG de contrôle avec HPN prolongée à 3 – 4 mois puis à 12 mois (23). Dans notre étude, les premiers EEG demandés montraient une nette amélioration des tracés dans 78% des cas pour l’EAE et 81% pour EAA. Les EEG anormaux concernaient les patients associant d’autres types de crises ou de troubles. Dans la littérature, les troubles cognitifs sont de plus en plus rapportés, en relation avec l’EA. Il s’agit souvent de troubles du langage, de la mémoire et attentionnels et parfois de troubles du comportement (5,22). Bulteau C. rapportait chez des patients avec EAE non traités, 35 % de déficit attentionnel, et 30 à 50 % d’hyperactivité (5). Dans notre cohorte, nous avions une proportion de 22,6 % de troubles de la mémoire. L’âge de début précoce des crises interférait dans la survenue des troubles cognitifs, dans la littérature (12,22) et dans notre étude avec une réduction significative des troubles mnésiques (23,8 % des EAE et 18,2 % des EAA). Un délai diagnostique et donc thérapeutique tardif et la survenue de CGTC constituent des éléments de mauvais pronostic pour la cognition des patients dans les EA (23). Ainsi, la recherche systématique de troubles neuropsychologiques associés par des tests appropriés doit être la règle car ces troubles peuvent persister, même après la régression des crises d’absence, et représenter un handicap pour la scolarité des patients, leur avenir psycho-intellectuel et conduire à des troubles psycho-sociaux à long terme (5,21). CONCLUSION Cette étude menée dans le contexte neurologique et neuropédiatrique sénégalais nous a permis de mettre en évidence une prévalence relativement élevée des épilepsie-absences chez l’enfant et l’adolescent. Leurs étiologies restent multifactorielles mais devant le taux élevé de consanguinité parentale observé dans ce contexte sénégalais nous craignont qu’elles soient principalement génétiques. Cependant leur diagnostic est relativement aisé du point de vue clinique et paraclinique. Dans la majorité de nos cas, les absences sont les seuls types de crises observées et elles répondent bien au traitement par valproate de sodium, éthosuximide, mais aussi à d’autres types de traitements. L’épilepsie-absence disparaît bien souvent avant l’âge adulte dans la majorité des cas sans séquelles, même si quelques troubles cognitifs ont été retrouvés chez certains enfants avec un impact sur leur scolarisation.
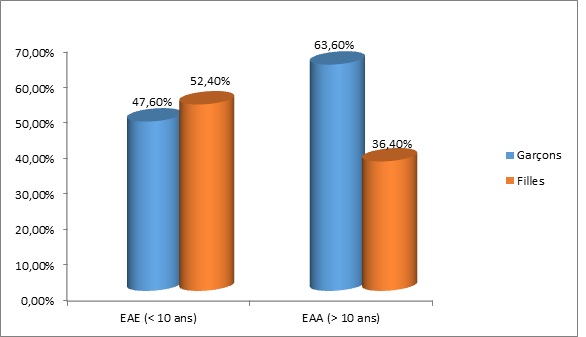 Figure 1 : Répartition des patients suivant le type d’absence et le sexe
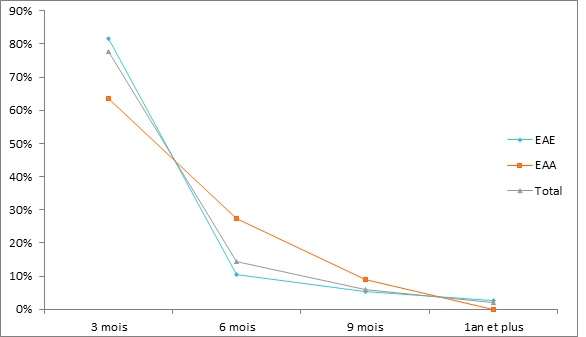 Figure 2: Répartition des patients suivant les délais d’arrêt des crises sous traitement par VPA
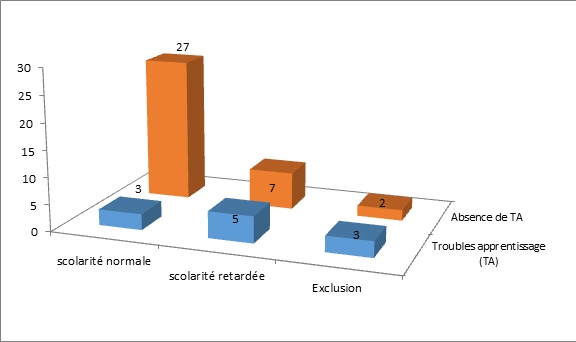 Figure 3 : Scolarité et troubles d’apprentissage
Tableau 1: Tableau récapitulatif des données cliniques descriptives des crises suivant le type d’absence
(Caractéristiques de l’épilepsie-absence chez l’enfant et l’adolescent : facteurs déclenchant, nombre de crise par jour, durée de la crise, signes associés) REFERENCES :
|
© 2002-2018 African Journal of Neurological Sciences.
All rights reserved. Terms of use.
Tous droits réservés. Termes d'Utilisation.
ISSN: 1992-2647