
|
|||||||||||||||||||||||||||||||||||||
|
CASE REPORT / CAS CLINIQUE
LA MYASTHENIE DE L’ENFANT : A PROPOS DE 5 CAS OBSERVES A ABIDJAN EN COTE D’IVOIRE
MYASTHENIA IN CHILDREN: ABOUT 5 CASES OBSERVED IN ABIDJAN, IVORY COAST
E-Mail Contact - DIAKITE Ismaïla :
di.smael@yahoo.fr
RESUME Introduction : La myasthénie est une maladie auto-immune rare chez l’enfant. Elle est peu décrite dans la littérature africaine car encore méconnue. Méthodologie : Nous rapportons 5 observations de myasthénie chez des enfants âgés de 0 à 15 ans répertoriés en 3ans d’Octobre 2015 à Décembre 2018 en consultation de neuropédiatrie et d’ophtalmologie à Abidjan. Le diagnostic de myasthénie a été posé sur des bases cliniques et/ou électroneuromyographiques (ENMG) et sur les dosages d’anticorps spécifiques. Résultats : L’âge moyen des patients était de 5,8 ans. Le sex ratio était de 1,5. Le ptosis était le signe révélateur dans tous les cas. Le test au glaçon ainsi que les anticorps anti RaCh (récepteur de l’acétylcholine) avaient été réalisés chez 3 patients. L’ENMG a montré un décrément significatif de plus de 10% chez tous les patients. Un seul patient avait une atteinte thymique. Tous nos patients ont reçu des anticholinestérasiques, 40% des corticoïdes et 20% l’azathioprine. L’évolution s’est faite vers une amélioration chez 40% (2/5) des patients, une aggravation chez 20% (1/5) et le décès chez 2 patients (40%). Le délai moyen du diagnostic était de 13 mois. Conclusion : L’étude montre un retard et une errance diagnostique. Elle suggère d’évoquer la myasthénie devant tout ptosis uni ou bilatéral, asymétrique et/ou fluctuant, chez l’enfant et de réaliser un test au glaçon, ainsi que, dans la mesure du possible, les tests aux anticholinestérasiques et le dosage des Ac anti-RaCh. Mots clés : Afrique- Anticholinestérasique- Enfant- Myasthénie- Ptosis- Récepteur de l’Acétylcholine SUMMARY Background: Myasthenia is a rare autoimmune disease in children that is poorly described in African literature. Method: We reported 5 cases of myasthenia gravis in children aged from 0 to 15 years observed in a 3 years period (October 2015 to December 2018) at outpatient’s child neurology consultation and ophthalmology in Abidjan, Ivory Coast. the diagnosis of myasthenia gravis was made on the basis of clinical, electroneuromyographic and immunological signs (anti-achR antibodies). Results: The mean age of patients was 5.8 years. The sex ratio was 1.5. Ptosis was the common presentation in all cases. The ice pack test and anti-AChR antibodies were performed in 3 patients. ENMG showed a significant decrement of more than 10% in all patients. Only one patient had thymic involvement. All patients received anticholinesterase drugs. As adjunctive treatment, 2 patients received steroids and one patient received azathioprine. There was an improvement in 40% (2/5) of the patients, a deterioration in 20% (1/5) and death occurred in 2 patients (40%). The average time to diagnosis was 13 months. Conclusion: This study shows a delay and wandering in the diagnosis. We recommend that myasthenia gravis should be suspected in the presence of any single or bilateral, asymmetrical and / or fluctuating ptosis in children. Moreover, whenever possible, an ice pack and pharmacological tests should be performed, and anti-achR dosage levels as well. Key words: Myasthenia gravis- Child- Ptosis- Acetylcholine receptor -Anticholinesterase – Africa INTRODUCTION : La myasthénie est une maladie auto-immune de la jonction neuromusculaire. Son évolution est souvent capricieuse, marquée fréquemment par des poussées alternant avec des phases de rémission. Les poussées peuvent être provoquées par certains facteurs déclenchants. L’atteinte des muscles respiratoires en fait toute la gravité. La prise en charge précoce et parfois multidisciplinaire et la palette des traitements actuels ont considérablement réduit la mortalité de cette affection avec l’utilisation des immunoglobulines et les échanges plasmatiques (2). La myasthénie de l’enfant est rare (13). Elle représente 10 à 15% des cas de myasthénie chez les caucasiens (15). En Asie particulièrement en Chine, cette fréquence peut atteindre 50% dans la présentation oculaire (20). En Afrique subsaharienne peu de données sont disponibles sur la myasthénie de l’enfant. Deux cas ont été rapportés au Togo (14) et 9 cas au Sénégal (1). En côte d’Ivoire, aucune étude n’a concerné la myasthénie de l’enfant. Notre objectif était de décrire à partir de 5 observations le tableau clinique, et l’évolution sous traitement de la myasthénie chez l’enfant et faire une revue de la littérature sur le traitement. MATERIEL ET METHODES : Nous avons réalisé une étude rétrospective, dans les unités de consultation de neuropédiatrie du CHU de Yopougon et de l’hôpital Mère-Enfant de Bingerville, dans l’unité de consultation du service d’ophtalmologie du CHU de Treichville à Abidjan. Tous les dossiers d’enfants âgés de 0 à 15 ans, chez qui le diagnostic de myasthénie a été posé sur des signes cliniques et/ou ENMG et sur les dosages d’anticorps spécifiques ont été colligés, durant une période de 3 ans, d’Octobre 2015 à Décembre 2018. OBSERVATIONS Observation n°1 : K.M, de sexe masculin, âgé de 14 mois, a été reçu en ophtalmologie en 2015 pour un ptosis bilatéral installé depuis un mois auparavant, d’abord à droite puis à gauche avec un strabisme divergent de l’œil gauche. L’examen physique notait un bon état général, un ptosis bilatéral asymétrique prédominant à droite, maximal en fin de journée. Le test au glaçon était positif. Les tests pharmacologiques n’ont pas été réalisés. Le dosage du taux des anticorps anti-récepteurs à l’Ach et le scanner thoracique n’ont pas été fait. L’ENMG a montré un décrément significatif (fig 1). Le patient a été traité par le chlorure d’ambénonium (Mytelase®). L’évolution a été marquée par une généralisation de la myasthénie, suivie du décès du patient dans un contexte de détresse respiratoire malgré l’intubation et la ventilation en réanimation, 13 mois après la consultation. Le traitement par les immunoglobulines n’a pu être réalisé. Observation n°2 : B.F., de sexe féminin, âgée de 02 ans et 07 mois, a été reçue en neuropédiatrie en 2018, pour un ptosis unilatéral gauche évoluant depuis un mois avec des chutes fréquentes. Ses antécédents personnels étaient marqués par un retard de langage, une non-acquisition de la propreté sphinctérienne et une alimentation faite uniquement de liquides. Elle n’avait pas d’antécédent familial particulier. L’examen physique notait un bon état général, un ptosis unilatéral maximal en fin de journée. Le test au glaçon était positif. Le reste de l’examen neurologique était sans particularité. Les explorations paracliniques ont mis en évidence : un décrément de 10% à l’ENMG, un taux élevé d’Ac anti-récepteurs à l’Ach et une hyperplasie thymique sans processus tumoral individualisable, prédominant à droite au scanner cervico-thoracique (Fig 2). La thymectomie proposée a été refusée par la famille. Sous Mytelase®, l’évolution a été marquée par des épisodes de poussées motivant l’augmentation de la dose de Mytelase et l’association à la corticothérapie. Observation n°3 : K.G, de sexe masculin, âgé de 06 ans, sans antécédent particulier, a été adressé à la consultation de neuropédiatrie en 2017 pour un ptosis fluctuant à bascule, une paralysie de l’abduction de l’œil droit et une chute de la tête évoluant depuis deux mois. Des occlusions oculaires par pansements ont initialement été réalisées sans succès. Secondairement, est apparu le ptosis gauche s’aggravant dans la journée. Le tonus musculaire était normal. Il n’y avait pas d’atteinte d’autres groupes musculaires. Le test au glaçon et les épreuves pharmacologiques n’ont pas été réalisés. L’ENMG objectivait un décrément significatif et le dosage des AC anti-récepteur à l’Ach était positif avec un taux égal à 0,4 nmol/l. Le patient a été traité par la Pyridostigmine ou Mestinon® avec une évolution stationnaire. Observation n°4 : K.Y., sexe masculin, âgé de 08 ans, reçu en 2017, a présenté plus d’un mois avant sa consultation en neuropédiatrie, une fatigabilité à la marche avec réduction du périmètre de marche, sans douleur ; un ptosis fluctuant, une anorexie, une voix nasillarde et une perte de poids d’environ 3 kg en moins d’un mois. Aucun antécédent familial n’a pu être noté. L’examen physique notait un bon état général, un ptosis bilatéral, le test au glaçon et les épreuves pharmacologiques n’ont pas été réalisés. Un décrément significatif a été objectivé par l’ENMG. Le bilan biologique thyroïdien et le scanner thoracique étaient normaux. Le dosage des Ac AntiAch n’a pu être réalisé. Sous chlorure d’ambénonium (Mytelase®) associé à la corticothérapie, l’évolution a été d’abord marquée par une amélioration de la symptomatologie, puis par le décès dans un contexte de généralisation de la myasthénie puis une détresse respiratoire malgré l’intubation et la ventilation en réanimation. Le traitement par les immunoglobulines et/ou l’échange plasmatique n’a pu être réalisé. Observation n°5 : T.M., de sexe féminin, âgée de 12 ans et 09 mois, sans antécédent personnel et familial particulier, a consulté en neuropédiatrie en 2018 pour un ptosis droit invariable au cours de la journée, parfois présent dès le matin au réveil. Une tarsorraphie a été réalisée sans succès. Depuis 5 ans, un ptosis gauche est apparu. Le test au glaçon était positif ; les tests pharmacologiques n’ont pas été réalisés et le dosage des Ac antiAch R était positif avec un taux de 10,8 nmol/L. La patiente a été traitée par la Pyridostigmine (Mestinon®) et l’Azathioprine (Imurel® 25 mg). L’évolution sous traitement a été satisfaisante. DISCUSSION Nous avons recensé 05 cas de myasthénie de l’enfant sur une période de 3 ans. Au Sénégal, sur une période de 5 ans, Basse et al. ont recensé 09 cas de myasthénie chez l’enfant (1). Au Brésil, Morita et al. avaient observé 18 cas de myasthénie de l’enfant sur une période de 14 ans (12). Malgré nos biais de recrutement, toutes ces données semblent confirmer la rareté de la myasthénie chez l’enfant. En Asie, la myasthénie de l’enfant est plus fréquente (20). En Chine, sur une population de 391 myasthéniques, Xiaofan et al ont trouvé 50% d’enfants (19). Aucune donnée de la littérature n’explique clairement cette rareté de la myasthénie de l’enfant. L’âge de nos patients, lors du diagnostic, variait de 01 à 12 ans avec un âge moyen qui était de 5,8 ans. La série sénégalaise situait l’âge moyen de leurs patients au moment du diagnostic à 6,2 ans (1). Vander Pluym et al. ont trouvé un âge de début des symptômes qui se situait entre 18 mois et 11 ans et avant 3 ans dans 8/18 des cas) (18). Deux (2) enfants étaient de sexe féminin, et 3 de sexe masculin. Selon Finnis, à l’âge pré-pubertaire, il y a une égalité au niveau des deux sexes dans la survenue de la myasthénie (6). Comme dans les séries togolaise (14) et sénégalaise (1), les signes inauguraux étaient oculaires représentés par un ptosis sans diplopie. Un seul patient avait des signes bulbaires et spinaux associés au ptosis. Selon Grob et al., classiquement, tous âges confondus, la symptomatologie initiale de la myasthénie est une atteinte oculaire dans 60 % des cas (9). Cette assertion est aussi vérifiée chez l’adulte, comme le démontre l’étude de Gnonlonfoun et al. qui a rapporté un ptosis et/ou une diplopie chez tous leurs patients (8). Le ptosis étant quasi-présent dans les myasthénies juvéniles, il serait souhaitable de réaliser le test au glaçon devant tout ptosis, même si sa positivité ne traduit pas exclusivement la présence d’une myasthénie. Un trouble de la phonation associé à une atteinte oculaire a été retrouvé chez un seul patient. Une atteinte de la musculature spinale, se manifestant par des troubles de la marche ou des difficultés à tenir la tête, était présente chez un seul patient au début de la maladie. Selon Grob et al., 20 % des myasthénies débutent par une atteinte bulbaire ou faciale et 20% des myasthénies débutent par un déficit de la musculature axiale ou périphérique (9). Le délai diagnostique moyen était de 13 mois, avec des extrêmes de 01mois et de 5 ans. Nidain et al. ont rapporté des délais diagnostiques de 6 et 9 ans chez deux sœurs au Togo (14) Ce long délai pourrait s’expliquer par la méconnaissance de la maladie, d’où une errance diagnostique chez différents professionnels de la santé avec un retard au diagnostic ; au cours de cette errance l’une de nos patientes avait même bénéficié d’une tarsorraphie sans succès. Le test au glaçon a été réalisé chez 3 patients sur 5. L’électroneuromyographie (ENMG) avec la technique des stimulations répétitives à basse fréquence de 3Hz en objectivant un décrément significatif a été contributif au diagnostic. L’étude du jitter lors de l’examen en fibre unique n’est pas utilisée en routine (5). Dans notre contexte, le dosage des anticorps est encore très souvent inaccessible dans les hôpitaux publics. Nous avons pu mettre en évidence des anticorps anti-RAch chez 3 patients (60%). La série sénégalaise a retrouvé des Ac anti-RAch dans 77,77% des cas (1). Pour Xiaofan et al., 66% des adultes et 64% des enfants atteints de myasthénie avaient des anticorps anti-RAch (19). Sur nos cinq patients, deux (40%) avaient réalisé un scanner thoracique, l’hyperplasie thymique a été mis en évidence chez un seul. Dans la série du Sénégal, un seul patient avait une atteinte thymique (11,11%) (1). En France, Boumendil et al ont observé une atteinte thymique dans 15 à 20% des cas (3), alors qu’aucune anomalie thymique n’a été rapportée par Gorafalo et al. à Cuba (7). Un thymome est retrouvé chez 10 à 15% des patients atteints de myasthénie avec un pic à l’âge de 50 ans et quel que soit le sexe (17). Les thymomes sont très rares, mais existent dès l’âge de 4 ans. La thymectomie semble améliorer le taux de rémission (4). Parmi les autres affections auto-immunes associées à la myasthénie et qui doivent être recherchées systématiquement, les dysthyroïdies (maladie de Basedow, thyroïdites) sont les plus fréquentes et sont retrouvées chez 5 à 10% des patients (16). Nous n’avons pas objectivé d’affection auto-immune associée à la myasthénie. Par contre, chez l’adulte, Gnonlonfoun et al. ont mis en évidence une hyperthyroïdie chez un de leurs patients (16,67%) (8). Tous nos patients étaient traités par des anticholinestérasiques, 40% par des corticoïdes, et 20% par l’azathioprine. Aucun patient n’a bénéficié d’un traitement par les immunoglobulines intraveineuses ni par du rituximab en raison du coût prohibitif dans notre contexte de travail. Dans la série sénégalaise tous les patients étaient sous anticholinestérasiques et seulement 22,22% sous corticoïdes dès le début de la maladie (1). Dans la série brésilienne, tous les patients ont été traités en première intention par la pyridostigmine. Et pour les mêmes auteurs, le traitement de la myasthénie chez l’enfant se fait selon la sévérité de l’atteinte, à la recherche d’une rémission complète des symptômes (12). Selon la littérature, les anticholinestérasiques sont utilisés en première intention (10). S’ils sont efficaces dans 50 à 70% des cas sur un ptosis isolé, ils restent le plus souvent insuffisants lorsque des troubles oculomoteurs sont présents, et ne dépassant pas 20% d’efficacité. Chez ces patients, non répondeurs aux anticholinestérasiques, les immunosuppresseurs sont plus efficaces. L’azathioprine reste le premier choix pour un traitement immunosuppresseur à long terme (11). La corticothérapie est généralement utilisée lorsque les symptômes ne sont pas suffisamment contrôlés par les anticholinestérasiques, Les résultats sont plutôt bons, puisque chez 72 à 92 % des patients traités, elle a entrainé une rémission ou une amélioration importante (17). Rarement utilisé seul du fait de son long délai d’action, l’azathioprine est actuellement souvent utilisée en association avec les corticoïdes (17) avec de très bons résultats. Cette association permettrait de consolider une rémission mais également de réduire la posologie des corticoïdes limitant ainsi leurs effets secondaires (5). Les immunoglobulines et les échanges plasmatiques, le rituximab sont actuellement disponibles dans notre contexte. Mais l’accès aux immunoglobulines ou aux échanges plasmatiques ou au ritixumab reste encore difficile. L’indication de ces médicaments suscités a été posé chez deux patients, le coût a été un frein. L’évolution sous traitement était marquée par une amélioration chez deux de nos patients. Chez un patient, nous avons noté une aggravation qui a motivé l’augmentation des doses d’anticholinestérasiques. Le décès est survenu chez deux autres patients après généralisation de la myasthénie et dans un contexte de détresse respiratoire, malgré l’association d’une corticothérapie. Il apparait donc nécessaire de surveiller les formes oculaires chez l’enfant même s’il est admis que la myasthénie du sujet âgé semble se généraliser plus fréquemment que chez le sujet jeune (5). La série sénégalaise a noté une amélioration nette chez 4 patients sur 9 et la survenue de 2 décès dans un tableau d’insuffisance respiratoire aigüe (1). Au Togo, les patientes ont été perdus de vue (5). Sur tous les patients de Morita traités à la pyridostigmine, il y a eu une rémission complète chez deux patients (12,5%) et trois patients (18,7%) ont présenté une amélioration clinique ; l’association de la prednisone chez 9 patients n’a pas entrainé de rémission complète, mais une stabilité chez 5 patients (50%) (12). En Chine, plusieurs enfants ont reçu de la prednisone sans rémission clinique évidente (19). Les formes oculaires de la myasthénie sont l’apanage de l’enfant à l’âge pré-pubertaire, avec une évolution vers la myasthénie généralisée à l’âge pubertaire. La rémission spontanée ou après traitement s’observe beaucoup plus à l’âge pré-pubertaire (6). CONCLUSION L’analyse des 5 observations de myasthénie chez l’enfant, a montré qu’elle a un début oculaire (ptosis), dans la quasi-totalité des cas. L’errance diagnostique est fréquente dans notre contexte de travail liée à la méconnaissance de l’affection. Il convient donc d’évoquer la myasthénie devant tout ptosis uni ou bilatéral, asymétrique et/ou fluctuant, chez l’enfant et de réaliser un test au glaçon, ainsi que, dans la mesure du possible, les tests aux anticholinestérasiques et le dosage des Ac anti-RAch. La séropositivité d’anticorps anti-récepteurs à l’acétylcholine est fréquente et souvent associée à une tumeur de thymus à rechercher obligatoirement. L’évolution est comparable à celle de l’adulte car la généralisation suivie du décès n’est pas rare. Le traitement est basé sur les anticholinestérasiques qu’il faut associer à la corticothérapie et/ou à l’azathioprine en cas de non réponse aux anticholinergiques. La plasmaphérèse, les Immunoglobulines et le rituximab ont révolutionné le pronostic de cette maladie, mais ces moyens thérapeutiques restent inaccessibles dans notre contexte en raison de leur coût. Une subvention de ces médicaments par nos états pourrait améliorer considérablement la prise en charge.
 Figure 1: Décrément à l’ENMG
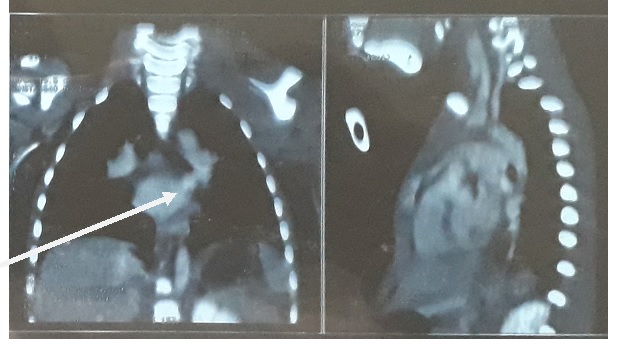 Figure 2: hyperplasie thymique au scanner cervico-thoracique.
Tableau I : Répartition des patients en fonction de l’âge et du sexe
Tableau II : Manifestations cliniques initiales et évolution sous traitement
REFERENCES
|
© 2002-2018 African Journal of Neurological Sciences.
All rights reserved. Terms of use.
Tous droits réservés. Termes d'Utilisation.
ISSN: 1992-2647