
|
|
|
CLINICAL STUDIES / ETUDES CLINIQUES
L’ATROPHIE MULTISYSTEMATISEE A LA CLINIQUE NEUROLOGIQUE IBRAHIMA PIERRE NDIAYE DE DAKAR, SENEGAL.
MULTIPLE SYSTEM ATROPHY AT THE IBRAHIMA PIERRE NDIAYE NEUROLOGICAL CLINIC, DAKAR, SENEGAL
E-Mail Contact - NGOMA Hermann Christel Kiendolo :
hermanngoma27@gmail.com
RESUME Introduction L’atrophie multisystématisée (AMS) est une affection neurodégénérative d’évolution progressive et fatale. De cause inconnue, elle est classée en deux formes cliniques (AMS-P et AMS-C). Patients et Méthode Nous avons réalisé une étude transversale portant sur neuf patients, membres de sept familles, répondant aux critères diagnostiques consensuels de l’AMS. Résultats L’âge moyen de survenue était de 43,7 ans. Les syndromes parkinsonien et cérébelleux ont été retrouvés dans 5 cas, un syndrome pyramidal dans 4 cas et une dysautonomie dans 3 cas. Une dysarthrie était présente dans 5 cas, des troubles cognitifs et une dysphagie dans 4 cas, des mouvements anormaux dans 2 cas, des troubles du sommeil et une hypoacousie chez 1 cas. La forme AMS-P était prédominante dans 5 cas, la forme AMS-C a été retrouvée chez 4 cas. L’imagerie cérébrale a objectivé dans 100% des cas une atrophie cérébelleuse et une atrophie du tronc cérébral. L’ENMG était en faveur d’une polyneuropathie chez trois patients et une atteinte du VIII a été objectivée chez un patient. Les pedigrees des quatre familles ont mis en évidence un mode de transmission autosomique récessive. Conclusion L’AMS considérée comme sporadique, peut aussi revêtir une caractéristique génétique. Nous avons rapporté des cas d’AMS chez des personnes issues de mariages consanguins, dont certaines ont des cas similaires dans la famille avec une prédominance de la forme AMS-P. Une cause génétique avec un mode de transmission autosomique récessive est fortement suspectée. Une étude génétique s’impose afin d’isoler les gènes. Mots clés : Atrophie multisystématisée, Consanguinité, Dakar, Sénégal. ABSTRACT Background Multiple system atrophy (MSA) is a progressive and fatal neurodegenerative disease. Of unknown cause, it is classified into two clinical forms (AMS-P and AMS-C). Patients and Method We carried out a cross-sectional study of nine patients, members of seven families, meeting the consensus diagnostic criteria of the AMS. Results The mean age of onset was 43.7 years. Parkinsonian and cerebellar syndromes were found in 5 cases, pyramidal syndrome in 4 cases and dysautonomia in 3 cases. Dysarthria was present in 5 cases, cognitive impairment and dysphagia in 4 cases, abnormal movements in 2 cases, sleep disturbances and hearing loss in 1 case. The AMS-P form was predominant in 5 cases, the AMS-C form was found in 4 cases. Brain imaging demonstrated cerebellar atrophy and brainstem atrophy in 100% of cases. The ENMG was in favor of polyneuropathy in three patients and VIII involvement was demonstrated in one patient. The pedigrees of the four families showed an autosomal recessive mode of inheritance. Conclusion AMS, considered to be sporadic, can also have a genetic characteristic. We have reported cases of AMS in people from consanguineous marriages, some of whom have similar cases in the family with a predominance of the AMS-P form. A genetic cause with an autosomal recessive mode of inheritance is strongly suspected. Genetic study is needed in order to isolate the genes. Key words: Consanguinity, Dakar, Multiple System Atrophy, Senegal. INTRODUCTION L’atrophie multi systématisée (AMS) est une affection neurodégénérative de l’adulte, d’évolution progressive et de pronostic sévère. Anciennement appelée syndrome de Shy Drager ou dégénérescence nigro-striée (DNS) ou atrophie olivo-ponto-cérébelleuse (AOPC), elle est considérée comme le syndrome parkinsonien atypique le plus fréquent (6). Elle est caractérisée par la combinaison variable d’un syndrome parkinsonien atypique, d’un syndrome cérébelleux, d’un syndrome pyramidal et d’une dysautonomie. Selon la prédominance des signes moteurs, elle est classée principalement en deux formes cliniques: l’AMS de type parkinsonien (AMS-P) et l’AMS de type cérébelleux (AMS-C) (22). Les critères diagnostiques de l’AMS définis en 1998 par Gilman et al., révisés en 2008 définissent trois niveaux de l’AMS : ‘’possible’’, ‘’probable’’ et ‘’certain’’. La certitude diagnostique est obtenue à l’examen neuro-pathologique post mortem, montrant une dégénérescence des structures olivo-ponto-cérébelleuses et de la voie nigro-striée associée à d’abondantes inclusions gliales intra cytoplasmiques d’alpha-synucléine. La cause exacte de l’AMS n’est pas encore connue et il n’existe toujours pas de traitement spécifique (7,8,20). La présence d’antécédents familiaux constituait jadis, un critère d’exclusion de l’AMS, qui était à l’époque considérée sans base génétique. Toutefois, des cas familiaux d’AMS ont été rapportés en Allemagne, aux Etats-Unis, en Chine, au Japon et au Sénégal (14,15,16,20). Ces études ont relancé l’hypothèse d’une part génétique de l’AMS mais les mutations génétiques n’ont pas encore été formellement identifiées. PATIENTS ET METHODE L’étude s’est déroulée à la clinique neurologique Ibrahima Pierre Ndiaye du Centre Hospitalier National Uiversitaire de Fann à Dakar, Sénégal. L’objectif était de décrire les caractéristiques sociodémographiques, cliniques et paracliniques des patients, et réaliser le phénotypage. Il s’est agi d’une étude transversale, réalisée du 1er décembre 2015 au 31 mai 2017, portant sur 9 patients, membres de 7 familles. Toute personne diagnostiquée AMS selon les critères consensuels révisés en 2008 (11) a été incluse dans l’étude. Tout patient qui ne répondait pas aux critères diagnostiques cliniques de l’AMS a été exclu. Les données ont été recueillies à l’aide d’un questionnaire qui permettait de noter les caractéristiques sociodémographiques du patient, l’histoire de la maladie, les antécédents personnels et familiaux, l’examen clinique, paraclinique et l’évolution. Les patients au cours de l’évolution ont été réévalués en utilisant l’échelle MDS-UPDRS (12). Les patients ont bénéficié d’une évaluation neuropsychologique avec le Test du Sénégal (TDS) (30). Le consentement éclairé et libre de tous les patients et/ou de leurs parents a été sollicité et obtenu avant toute soumission de questionnaire. RESULTATS L’âge moyen des patients était de 43,7 ans avec des extrêmes de 23 à 62 ans. La distribution était bimodale avec des tranches d’âge de 20-35 ans et 51-64 ans. Cinq patients étaient originaires de Dakar, deux patients provenaient de Thiès et deux autres de Louga. Sept patients sur neuf (77,8 %) étaient nés d’un mariage consanguin (consanguinité de 2ème degré). Parmi les sept, six avaient des cas similaires dans la famille dont trois étaient issus de la même famille (frères et sœurs utérins mais aussi germains). L’âge moyen des patients au début de la maladie était de 38,3 ans avec des extrêmes de 12 et 61 ans. Quatre patients présentaient un syndrome cérébelleux, quatre autres un syndrome parkinsonien et un patient présentait un syndrome parkinsonien prédominant associé à un syndrome cérébelleux statique tardif. La dysphagie était présente dans 44,4% des cas, la dysarthrie dans 55,5% et la dysautonomie dans 33,3% des cas. Des mouvements dystoniques du membre supérieur ont été notés (2 cas). L’hypoacousie a été retrouvée chez un patient. Les troubles du sommeil ont été notés dans 11,1% des cas. Les troubles cognitifs ont été retrouvés chez deux patients avec des scores du TDS à 25/39 et 27/39. La forme AMS-P était présente dans 55,6% des cas et la forme AMS-C dans 44,4%. L’imagerie cérébrale (l’IRM et TDM) a mis en évidence chez tous les patients une atrophie cérébelleuse associée à une atrophie du tronc cérébral (figures 1 et 2). Sur les coupes sagittales, l’aspect de l’atrophie cérébelleuse rappelait le « signe de la feuille de Nébédaye ou Never Die » (figure 3) et l’atrophie du tronc cérébral le « signe du sabre ». Des anomalies du cortex ont été aussi observées : une atrophie fronto-pariétale chez trois patients, une atrophie fronto-pariéto-temporale chez un patient, atrophie frontale chez un patient et pariétale également chez un seul patient. Deux patients ont présenté une atrophie cortico-sous corticale. L’ENMG était sans particularité chez six patients. Elle était en faveur d’une polyneuropathie sensitive distale, axonale, symétrique des quatre membres chez un patient, une polyneuropathie sensitive débutante chez le deuxième et une polyneuropathie sensitivomotrice proximo-distale symétrique des quatre membres prédominant aux membres inférieurs chez le troisième patient. Les potentiels évoqués auditifs ont mis en évidence chez un patient une atteinte tronculaire axono-myélinique du nerf auditif gauche ; le nerf auditif droit était sans particularité. Les pedigrees des quatre familles suggéraient un mode de transmission autosomique récessif (figure 4). Toutes ces familles avaient une notion de consanguinité parentale. L’évolution était stationnaire chez tous les autres patients. Sur le plan fonctionnel, avec l’échelle MDS-UPDRS à la section V, six patients sur neuf présentaient une dysautonomie fonctionnelle à la section V, au stade 4 de Hoehn et Yahr, et ont dû interrompre leur activité professionnelle. L’échelle de Schwab et England à la section VI du MDS-UPDRS était entre 70 et 90% pour l’ensemble des patients.
DISCUSSION Nous avons retrouvé un âge moyen de 43,7 ans, ce qui est conforme aux données de la littérature avec une tranche d’âge de 30 à 75 ans (17,21,33). Cependant nos extrêmes allant de 23 à 62 ans, notamment notre patient de 23 ans, remettent en question le fait que l’AMS ne pourrait se manifester avant 30 ans. L’AMS débute en moyenne vers 54-60 ans avec une survie moyenne de 6 à 9 ans (9,19,28). Dans notre travail, l’âge moyen de début de la maladie était de 38,3 ans avec des extrêmes de 12 et 61 ans. Cet âge moyen de début de la maladie est proche de celui retrouvé à Thiès par Moulid qui était de 32,8 ans (21). Sept patients sur neuf (77,8%) avaient une notion de consanguinité parentale, dont trois étaient issus de la même famille, dans notre étude. Les hommes étant touchés autant que les femmes et le fait que la consanguinité parentale semble favoriser, amplifier l’expression de la maladie oriente vers une transmission autosomique récessive. Moulid et al (21), ainsi que d’autres auteurs ont rapporté ce mode de transmission chez des cas familiaux (14,28). Des études récentes ont retrouvé une forte implication de l’hérédité dans l’AMS, remettant ainsi en cause son aspect sporadique exclusif (14,16,28). Al-Chalabi et al (2) ont rapporté que des variantes du gène SNCA de l’alpha synucléine pourraient être associées à l’AMS. Cette association serait plus forte dans l’AMS-C. Ces gènes pourraient faire la preuve de la composante héréditaire dans le développement de l’AMS. Des tests génétiques spécifiques ont été réalisés sur toutes les séquences du gène SNCA chez des cas familiaux d’AMS aux USA, et aucune mutation sur le gène de l’alpha synucléine n’a été trouvée (15). Aucun gène, ni mutation responsable de l’AMS n’a été identifié jusqu’à maintenant. Des facteurs de risque non spécifiés pourraient être associés à la susceptibilité génétique (16), des investigations supplémentaires, plus poussées s’imposent. La durée moyenne d’évolution de la maladie était de 6,3 ans avec des extrêmes de 1 à 16 ans d’évolution. Un syndrome parkinsonien a été noté chez cinq patients (55,5%), un syndrome cérébelleux chez également cinq patients (55,5%), un syndrome pyramidal chez quatre patients (44,4%) et une dysautonomie chez trois patients (33,3%). Toutefois, la dysautonomie n’a pas été recherchée de façon objective. Une dysautonomie symptomatique et précoce serait associée à un pronostic péjoratif. Elle pourrait aussi apparaitre quelques années après le début de la maladie. Des cas de dysautonomie tardive plus de quinze ans après le début de la maladie ont été rapportés (13,24). D’autres signes ont été objectivés chez nos patients tels que les troubles cognitifs, évalués avec le test du Sénégal (30), une dysphagie, une dysarthrie, une hypoacousie et une dystonie. Les troubles cognitifs ont été retrouvés chez deux de nos patients (22,2%) avec des scores du TDS à 25/39 et 27/39. Ils étaient initialement considérés comme critères d’exclusion de l’AMS (31). Certains auteurs ont décrit des troubles cognitifs chez des patients AMS ces dernières années, suggérant leur retrait des critères d’exclusion (4,21,26,27). Les troubles du sommeil étaient présents chez une patiente (11,1%) dans notre étude. Ce résultat est proche de celui retrouvé par Moulid (21). Dans la littérature, ils seraient présents dans plus de 80%, surtout dans les formes cérébelleuses (10,29,31). La dysphagie était observée chez quatre patients (44,4%) dans notre étude. Elle fait partie des critères additionnels de l’AMS (9,19). Nous avons retrouvé la dysarthrie chez cinq (55,5%) de nos patients, dysarthrie en rapport avec le syndrome cérébelleux. La dysarthrie cérébelleuse fait partie des critères consensuels pour le diagnostic de l’AMS probable (9). L’hypoacousie n’est presque pas décrite dans la littérature dans l’AMS. Récemment, des auteurs ont retrouvé une baisse d’acuité auditive chez des patients AMS-C comparés à des sujets contrôles (28). A Thiès, Moulid (21) a retrouvé une atteinte bilatérale du VIII chez une patiente. Dans notre série, nous avons observé l’hypoacousie chez un patient (11,1%). Cette baisse d’acuité auditive a été confirmée par le PEA qui a révélé une atteinte du VIII. La cause n’est pas encore connue, il pourrait s’agir toutefois d’une possible dégénérescence du système nerveux périphérique, ce qui ferait de l’AMS une atteinte centrale et périphérique. Des mouvements anormaux à type de dystonie ont été notés chez deux de nos patients (22,2%). Les mouvements anormaux, selon certains auteurs, sont plus ou moins fréquents surtout dans la forme AMS-P (19,23,25). Quatre patients (55,6%) sur neuf présentaient la forme AMS-P, qui est légèrement plus fréquente que la forme AMS-C retrouvée chez quatre patients (44,4%). La littérature rapporte la prédominance de l’AMS-P en Nouvelle-Zélande (13,34) alors que l’AMS-C serait plus fréquente au Japon (14,35) et à Thiès au Sénégal (21). Nous avons noté une atrophie du cervelet et du tronc cérébral à l’imagerie, tant à la TDM qu’à l’IRM cérébrale, chez tous les patients (100%). L’IRM cérébrale a en plus montré une atrophie des pédoncules cérébelleux moyens. Sur les coupes sagittales, nous avons observé le « signe de la feuille deNever Die » concernant l’atrophie cérébelleuse et le « signe du sabre » concernant l’atrophie du tronc cérébral. Nous n’avons pas par contre visualisé le signe de la croix pontique sur les séquences pondérées T2 et Flair. L’atrophie des pédoncules cérébelleux moyens, du pont ou du cervelet font partie des critères additionnels pour le diagnostic d’AMS « possible » (5,9,18,32). Nous avons également retrouvé une atrophie frontale chez un patient, une atrophie fronto-pariétale chez trois patients, une atrophie fronto-pariéto-temporale chez un patient, et également une atrophie cortico-sous corticale chez deux patients. Ces anomalies ne sont pas rapportées dans la littérature, mais certains auteurs les ont identifiées à l’autopsie, à l’examen anatomopathologique post-mortem (8,19). L’ENMG a été en faveur d’une polyneuropathie sensitive chez trois patients et une polyneuropathie sensitivomotrice chez un seul patient ; elle était normale chez les autres patients. Dans la littérature, des cas de neuropathies périphériques ont été décrits chez 40% des patients AMS qui avaient une diminution significative des amplitudes sensitives (1). Moulid a retrouvé, à Thiès, une polyneuropathie sensitive des quatre membres chez quatre patients (21). La cause de ces neuropathies est encore inconnue, certains auteurs évoquent l’hypothèse d’une possible dégénérescence du système nerveux périphérique (1,3). La durée moyenne d’évolution de la maladie est de 6,3 ans dans notre série. Cette évolution est conforme aux données de la littérature faisant état de 6 à 9 ans (3,24,37). Cependant les extrêmes dans notre travail vont de 1 à 16 ans, ce qui est assez bas. Certains auteurs ont retrouvé des survies pouvant dépasser vingt ans (22,27,33). CONCLUSION L’AMS est toujours considérée comme sporadique, et très peu de cas familiaux ont été rapportés dans le monde. Notre étude a rapporté des cas d’AMS chez des personnes issues de mariages consanguins, dont certaines ont des cas similaires dans la famille avec une prédominance de la forme AMS-P. A la vue des pedigrees de ces familles, l’hypothèse d’une étiologie génétique selon un mode de transmission autosomique récessive nous parait fort probable. Une étude génétique avec des techniques performantes de séquençage s’impose pour conforter notre hypothèse. Conflit d’intérêt : Aucun.
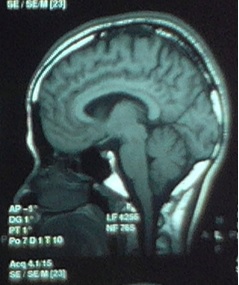 Figure 1 (Patient A.H.T) : IRMc coupe sagittale T1 montrant une atrophie modérée du tronc cérébral réalisant le « signe du sabre », une atrophie du cervelet (« signe de la feuille de Nébédaye ou Never Die ») et une atrophie du cortex pariétal.
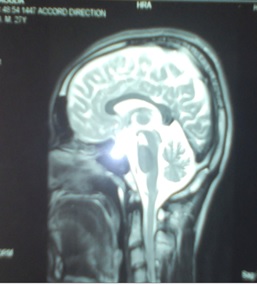 Figure 2 (Patient D.T) : IRMc coupe sagittale T2 mettant en évidence une atrophie sévère cérébelleuse (« signe de la feuille de Never Die ») et du tronc cérébral (« signe du sabre ») associées à une atrophie cortico-sous corticale.
 Figure 3 : Feuille de « Nébédaye ou Never Die ».
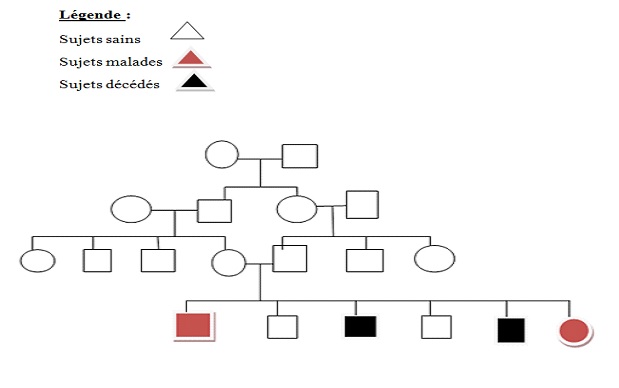 Figure 4 : Pedigree d’une famille REFERENCES
|
© 2002-2018 African Journal of Neurological Sciences.
All rights reserved. Terms of use.
Tous droits réservés. Termes d'Utilisation.
ISSN: 1992-2647