
|
|
|
CLINICAL STUDIES / ETUDES CLINIQUES
MANIFESTATIONS ELECTRO-CLINIQUES DU SYNDROME DE LENNOX –GASTAUT
ELECTRO-CLINICAL MANIFESTATIONS OF LENNOX–GASTAUT SYNDROME
E-Mail Contact - OUEDRAOGO Pingdéwendé Victor :
pvictoro123@gmail.com
RESUME Introduction : Le syndrome de Lennox-Gastaut (SLG) est une encéphalopathie épileptique rare. Notre étude consiste à déterminer les caractéristiques électro-cliniques du SLG au centre hospitalier national universitaire de Fann à DAKAR. Méthodologie: Il s’agit d’une étude rétrospective portant sur les dossiers médicaux de juillet 2004 à mai 2015, réalisée à la clinique neurologique et à l’hôpital des enfants Albert Royer. L’âge des enfants, l’âge de début des crises, les antécédents pathologiques, le type de crise, les troubles cognitifs et les tracés EEG ont été recueillis. Ont été inclus dans cette étude, tous les patients présentant de multiples types de crises, partielles ou généralisées ; un retard mental et à l’EEG, des pointes ondes lentes généralisées à la veille et/ou pointes rapides rythmiques pendant le sommeil. Résultats: Nous rapportons cinq cas de SLG de différents âges avec comme antécédents, une épilepsie familiale chez trois enfants et un syndrome de west chez un enfant. Le sex ratio était de 3/2 en faveur du sexe masculin. L’âge de début du SLG se situait typiquement entre 1 et 7 ans chez 4 patients. Les types de crises retrouvées étaient les crises toniques (100%), les absences atypiques (80%), les crises partielles motrices (60%), les crises atoniques (40%) et crises généralisées tonico-cloniques (40%). Aussi, tous les enfants avaient des troubles cognitifs. Les patterns EEG les plus fréquemment retrouvés étaient des décharges de Pointes ondes lentes Conclusion : Le SLG est une encéphalopathie épileptique infantile rare caractérisée par de multiples types de crise, un tracé EEG spécifique et un retard psychomoteur. Les résultats de notre étude concernant le profil électro-clinique se rapprochent de ceux de la littérature. Mots clés: Encéphalopathie épileptique – syndrome de Lennox-Gastaut – pointe-onde lente. ABSTRACT Introduction: Lennox–Gastaut syndrome (LGS) is an uncommon epileptic encephalopathy. In this study, we tried to determine the clinical and EEG characteristics of patients with LGS in DAKAR Fann teaching hospital. Methods: It is a retrospective study concerning medical folders during July 2004 to May 2015 realized in neurological clinic and Albert Royer children’s hospital. Age, gender, age at seizure onset, seizure type(s), epilepsy risk factors, cognitive disorders and EEG of all patients were registered. We included for this study all patients presenting multiple seizure types, mental retardation and an interictal EEG showing bursts of slow spike-wave complexes during awake and/or generalized paroxysmal fast activity during sleep. Results: We report different age five LGS cases with familial epilepsia in three cases and west syndrome in one case. The sex ratio was 3/2 for male. The age at seizure onset was typically between 1 and 7 years in four patients. The most common seizure type was tonic (100%), followed by atypic absence (80%), partial motor seizures (60%), and atonic seizures (40%) and generalized tonic–clonic seizures (40%). All the five childrens had cognitive disorders. The most common EEG finding was slow spike-wave complexes. Conclusion: LGS is an uncommon epileptic encephalopathy characterized by multiple seizure types, a specific electroencephalographic pattern and psychomotor retardation, beginning in childhood. Our results are comparable with literature. Keywords: Epileptic encephalopathy – Lennox-Gastaut syndrome –Slow spike wave. INTRODUCTION Le syndrome de Lennox-Gastaut est une encéphalopathie épileptique spécifique à un âge donné, caractérisée par des crises épileptiques multiples; des pointes-ondes lentes à l’électroencéphalogramme (EEG) de veille, des bouffées de rythmes rapides pendant le sommeil; un retard psychomoteur et des troubles de la personnalité [5]. L’encéphalopathie épileptique se réfère à un groupe hétérogène de conditions dans lesquelles même en absence d’anomalies cérébrales métaboliques et/ou structurelles progressives, l’activité épileptique (crises épileptiques et décharges à l’EEG) contribue à la détérioration cognitive et aux troubles du comportement [10]. L’incidence du syndrome de Lennox-Gastaut est très variable. Elle est estimée entre 1 à 10% de toutes les épilepsies de l’enfant et survient typiquement entre 1 et 7 ans [3]. Cette variabilité est dû au fait que certains auteurs utilisent des critères diagnostiques larges avec une tendance à considérer comme syndrome de Lennox-Gastaut, tous les patients ayant de multiples types de crises généralisées et une déficience intellectuelle [6]. Le diagnostic n’est pas toujours aisé dès lors que les manifestations électro-cliniques apparaissent progressivement. L’objectif général de cette étude était d’analyser les aspects électro-cliniques et évolutifs du syndrome de Lennox-Gastaut dans le service de neurologie et de pédiatrie du Centre Hospitalier National Universitaire (CHNU) de Fann de Dakar au Sénégal. Les objectifs spécifiques étaient d’une part de décrire les aspects cliniques des patients présentant un syndrome de Lennox-Gastaut et d’autre part, de décrire leurs aspects électroencéphalographiques. MATERIELS ET METHODES Il s’agissait d’une étude rétrospective réalisée à la clinique neurologique et à l’hôpital des enfants Albert Royer du Centre Hospitalier National Universitaire de fann de Dakar et portant sur les dossiers médicaux de juillet 2004 à mai 2015. Tous les patients présentant les critères électro-cliniques du SLG durant cette période ont été inclus. Ces critères comportaient de multiples types de crises, partielles ou généralisées; un retard mental et à l’EEG, des pointes ondes lentes généralisées à la veille et/ou pointes rapides rythmiques pendant le sommeil. Ont été exclus, tous les patients ne présentant pas de crises toniques ou ayant un dossier incomplet ou un suivi ambulatoire de moins d’une année. Les aspects anamnestiques, cliniques, électroencéphalograpiques ont été évalués grâce à une fiche établie à cet effet. RESULTATS Aspects cliniques : Durant la période d’étude, cinq enfants présentaient un syndrome de Lennox-Gastaut. Le sex ratio était de 3/2 en faveur du sexe masculin. L’âge de début du SLG se situait typiquement entre 1 et 7 ans chez 4 patients. L’âge minimal de début était d’un an alors que l’âge maximal était de 9 ans. Concernant les antécédents, notre étude a retrouvé une histoire familiale d’épilepsie chez trois patients et un syndrome de West (SW) chez un patient. Nous avons retrouvé deux types de crises chez un patient et au moins trois types de crises chez les quatre autres. Les types de crises retrouvés étaient les crises toniques (100%), les absences atypiques (80%), les crises partielles motrices (60%), les crises atoniques (40%) et crises généralisées tonico-cloniques (40%). Cependant, au cours de la consultation initiale, quatre patients présentaient un seul type de crise (crises toniques chez trois patients et crises généralisées tonico-cloniques chez un patient) et un seul patient présentait deux types de crises (crises toniques et crises partielles motrices). Tous les enfants avaient des troubles cognitifs : un déficit cognitif global chez trois patients, un trouble du langage chez deux patients et un syndrome autistique chez un patient. Cependant, un enfant âgé de 9 ans gardait une scolarisation normale. Aspects électroencéphalographiques : Des enregistrements électroencéphalographiques de veille (figures 1, 2, 4 ,6 et 7) ont été réalisés chez tous les enfants et des tracés de sommeil chez deux enfants (figures 3 et 5). Nous avons retrouvé des pointes-ondes lentes avec un ralentissement du rythme de fond chez tous nos cinq patients (100%) ainsi que des rythmes rapides au cours du sommeil chez deux patients (40%). DISCUSSION Le SLG est une encéphalopathie épileptique relativement rare. Il est le plus souvent caractérisé par une triade faite de plusieurs types de crises, un tracé EEG intercritique spécifique montrant des décharges de pointes-ondes lentes (≤2,5 Hz) et /ou des rythmes rapides et un retard mental. Notre étude rétrospective sur une période de plus de 10 ans a retrouvé 5 cas documentés. Une étude iranienne a retrouvé 135 cas de SLG sur une période de 4 ans [4]. Cette variabilité est due à la disparité des paramètres utilisés pour définir la maladie ainsi que la diversité des facteurs prédisposant parmi les différentes populations. Aussi, nous avons exclu plusieurs dossiers dont les tracés EEG n’étaient pas retrouvés dans nos archives. Notre étude a retrouvé un sex ratio de 3/2 en faveur du sexe masculin. Cette prédominance masculine est retrouvée dans d’autres séries [4, 12, 13, 14]. L’âge de début du SLG se situe typiquement entre 1 et 7 ans [2]. Dans la série d’Asadi-Pooya [4], l’âge moyen de début était de 3,2 ans. Dans l’étude de Goldsmith [11], l’âge moyen était de 4 ans. Ceci était analogue à notre étude où l’âge de début se situait entre 1 et 7 ans dans quatre de nos patients. Cependant, 15% des patients ayant un SLG ont un début tardif après huit ans [11] comme chez un de nos patients. Dans la thèse de Zouiri [14], 45% des patients avaient des antécédents pathologiques particuliers dont un syndrome de West dans 18,5% des cas. Goldsmith et al [11] ont retrouvé une histoire familiale d’épilepsie dans 13,1%, des convulsions fébriles dans 5,6%, et un retard de croissance dans 8,4%. Cela était similaire à notre étude qui a retrouvé un syndrome de west chez un patient et une histoire familiale d’épilepsie chez trois patients. Les manifestations cliniques du SLG sont plus ou moins similaires aux différentes études [1, 3, 6]. Ce syndrome est caractérisé par le polymorphisme des crises épileptiques et leur fréquence pluriquotidienne. Dans notre étude, au cours de la consultation initiale, le polymorphisme des crises épileptiques n’avait été retrouvé que chez un patient. Cependant, après au moins une année de suivi, nous avons retrouvé deux types de crises chez un patient et au moins trois types de crises chez quatre autres. Dans une série iranienne [4], 40% des patients avaient deux types de crises, tandis que 60% avaient au moins trois types de crises. Dans une série marocaine [14], on a rapporté 29,6% de crises toniques, 26,9% de crises atoniques et 29,6% d’absences atypiques. Goldsmith et al [11] ont montré que 77% des enfants avaient développé des crises toniques, 61% des absences atypiques et 56% des crises généralisées tonico-cloniques. Dans la série de Widdess-Walsh [13], les crises les plus communes étaient les absences atypiques (80 %), les crises atoniques (65,1 %), les crises généralisées tonico-cloniques (60 %) et des crises toniques (55,5 %). Dans notre étude le type de crise le plus fréquent était les crises toniques (100%), l’absence de crises toniques constituait un critère d’exclusion. Les autres types de crises retrouvés dans notre étude étaient les absences atypiques (80%), les crises partielles motrices (60%), les crises atoniques (40%) et les crises généralisées tonico-cloniques (40%). Cela est dû à la variabilité des critères diagnostiques utilisés par les différents auteurs et aux difficultés qu’ont les parents à reconnaître les différents types de crises. En effet, les crises toniques peuvent être subtiles et ne survenir que dans le sommeil. Aussi, les absences atypiques sont difficiles à diagnostiquer chez des enfants ayant une détérioration cognitive et comportementale et surtout qu’elles ont un début et une fin très progressifs et une rupture de contact incomplète. Les troubles cognitifs sont fréquents dans le SLG. Le retard psychomoteur et les symptômes psychiatriques surviennent dans 90% des cas. Le langage est le plus fréquemment affecté. Les troubles du comportement rapportés incluent l’hyperactivité, l’anxiété, l’agressivité, les traits autistiques ainsi que les troubles de la personnalité [13]. Dans notre étude, tous les enfants avaient des troubles cognitifs. Bien que le retard mental soit considéré comme un critère diagnostique du SLG, des auteurs ne l’incluent pas toujours dans leurs études et certains de leurs patients avaient un profil cognitif normal ou pseudo-normal [14, 24]. Dans une étude iranienne [4], 2,2% des patients avaient une fonction cognitive normale. Dans notre étude, un enfant poursuivait sa scolarisation. La signature EEG typique du SLG inclut un ralentissement du rythme du fond et des bouffées de pointes-ondes lentes qui peuvent être absentes au début de la maladie [9]. Les rythmes rapides paroxystiques généralisés avec un maximum antérieur peuvent être vus pendant le sommeil et sont associés à des crises toniques. Les patients avec un SLG ont un risque élevé de développer un état de mal non convulsivant pouvant être méconnu ou diagnostiqué tardivement. L’EEG posera le diagnostic en montrant des pointes-ondes lentes quasi-continues. Dans une étude iranienne [4], le ralentissement du rythme de fond était retrouvé dans 97,8% des cas et 86 ,7% des patients avaient des décharges de POL; les rythmes rapides étaient retrouvés dans 45,2% des cas. L’association décharges de POL et rythmes rapides représentait 37% des patients [4]. Ciccone et al [7] ont rapporté en Zambie le cas d’un garçon de 2 ans 7 mois suivi pour un SLG chez qui l’EEG du sommeil a montré une désorganisation du rythme de base, des décharges de POL de 1,5 à 2 Hz, des décharges généralisées de rythmes rapides ainsi que des pointes multifocales. Dans la série de Widdess-Walsh [13], les rythmes rapides ne représentaient que 12,6% des patients. Dans notre étude, 100% des patients avaient des décharges de POL et un ralentissement du rythme de fond. Cela est dû au fait que nous avions utilisé des critères stricts. Aussi, les rythmes rapides représentaient 40% de nos patients. Cela tient au fait que la vidéo-EEG de sommeil n’avait pas été réalisée de façon systématique chez tous nos patients. CONCLUSION Le SLG est une encéphalopathie épileptique rare débutant dans l’enfance, caractérisée par de multiples types de crises, un pattern EEG spécifique et un retard psychomoteur. Les résultats de notre étude concernant le profil électro-clinique se rapprochent de ceux de la littérature. L’apparition progressive des différents types de crises fait errer ou retarder le diagnostic. De même un début tardif et un bon développement psychomoteur sont retrouvés chez certains patients. Le diagnostic syndromique correct nécessite une analyse détaillée de l’histoire clinique. La répétition des examens électroencéphalographiques (veille + sommeil) chez tout cas d’encéphalopathie épileptique est indispensable pour poser précocement le diagnostic. FIGURES  Figure 1 : patient 1, 13 ans, EEG de veille avec une amplitude de 100 microvolts/cm (13/02/2013) Rythme de fond ralenti à 4-6Hz avec une décharge généralisée de POL à 2 Hz.
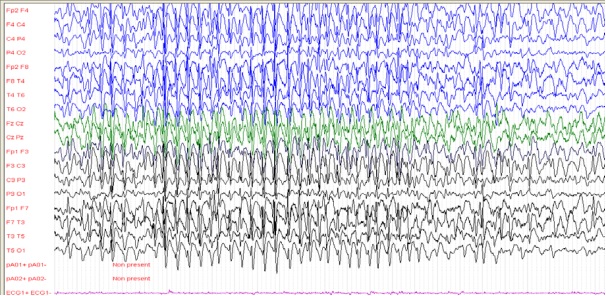 Figure 2: patient 2, 4 ans, EEG de veille avec une amplitude de 200 microvolts/cm (29/12/2005) Rythme de fond ralenti à 3-4 Hz avec plusieurs bouffées généralisées de POL à 2 Hz.
 Figure 3: patient 2, 4 ans, EEG de sommeil avec une amplitude de 200 microvolts/cm (29/12/2005) Plusieurs bouffées de rythmes rapides associées à des POL.
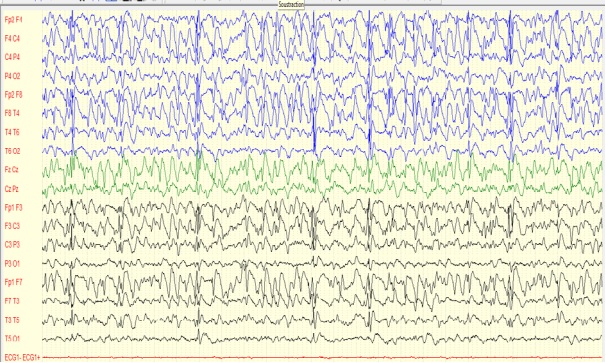 Figure 4: patient 3, 10 ans, EEG de veille avec une amplitude de 200 microvolts/cm (16/02/2006) Rythme de fond ralenti à 3 Hz avec plusieurs bouffées généralisées de POL à 2 Hz.
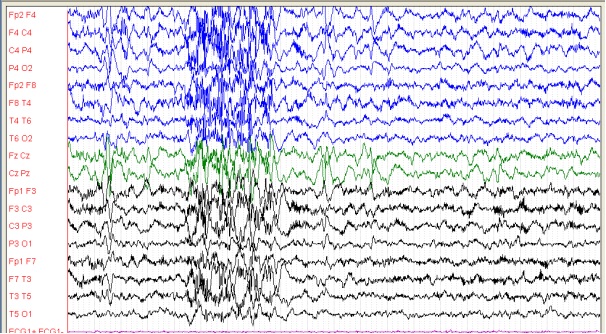 Figure 5: patient 4, 9 ans, EEG de sommeil avec une amplitude de 100 microvolts/cm (01/02/2013) Plusieurs bouffées diffuses de rythmes rapides associées à des pointes-ondes lentes.
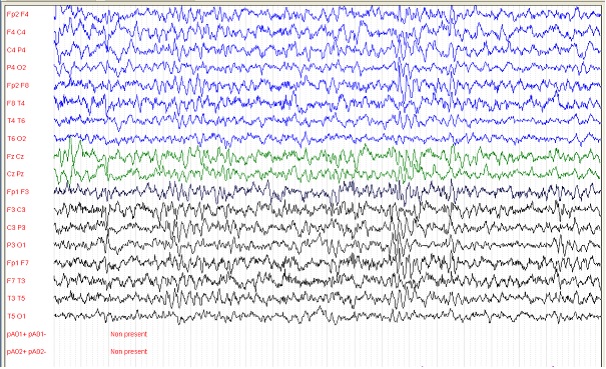 Figure 6: patient 4, 9 ans, EEG de veille avec une amplitude de 100 microvolts/cm (01/02/2013) Bouffées généralisées d’ondes lentes à 2-3 Hz souvent encochées de pointes.
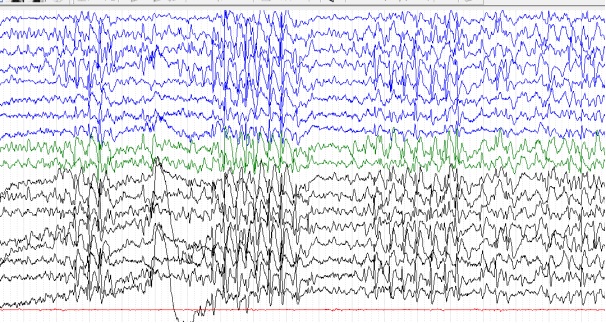 Figure 7: patient 5, 14 ans, EEG de veille avec une amplitude de 100 microvolts/cm (02/08/2013) Rythme de fond ralenti à 4-5 Hz avec plusieurs bouffées généralisées de POL à 2Hz REFERENCES
|
© 2002-2018 African Journal of Neurological Sciences.
All rights reserved. Terms of use.
Tous droits réservés. Termes d'Utilisation.
ISSN: 1992-2647