
|
|||||||||||||||||||||||||||||||||||||||||||
|
CLINICAL STUDIES / ETUDES CLINIQUES
PROFIL EPIDEMIOLOGIQUE DE L’EPILEPSIE CHEZ DES PATIENTS ATTEINTS DE TROUBLES DU SPECTRE DE L’AUTISME: ETUDE DE 45 CAS A DAKAR (SENEGAL)
EPIDEMIOLOGIC PROFILE OF EPILEPSY IN PATIENTS WITH AUTISM SPECTRUM DISORDERS: STUDY OF 45 CASES IN DAKAR (SENEGAL)
E-Mail Contact - SOW Adjaratou Dieynabou :
sowads@gmail.com
RESUME Description Troubles du spectre de l’autisme (TSA) et épilepsie peuvent coexister chez une même personne constituant des facteurs de mauvais pronostic bilatéraux. Objectif Décrire les éléments sociodémographiques de patients atteints de TSA et étudier les aspects clinico-paracliniques et évolutifs des épilepsies chez ces patients. Patients et Méthodes Etude transversale et descriptive au service pédopsychiatrique du CHNU Fann, avec analyse des dossiers, via un questionnaire standardisé, de tous les patients suivis entre Janvier 2004 et Septembre 2018 pour TSA avec ou sans épilepsie. Résultats Quarante-cinq patients avec TSA colligés, avec une fréquence de l’épilepsie de 37,8%. L’épilepsie avait débuté avant l’âge de 5 ans dans 94% des cas. Les crises étaient généralisées (58,8%), essentiellement motrices tonico-cloniques (80%), ou focales (35,3%), avec une fréquence de 2 crises/jour à 1 crise/semaine. L’EEG de veille et sommeil montrait des anomalies majoritairement frontales dans 60%, et centro-pariétales dans 26,6% des cas. L’imagerie cérébrale était normale dans 93,9% des cas, et les potentiels évoqués auditifs (PEA) normaux dans 87,2% des cas. La prise en charge était multidisciplinaire pour les TSA (neuropsychologique, psychomotrice, orthophonique) et médicale, essentiellement (82,4%) en monothérapie pour l’épilepsie. Les médicaments antiépileptiques utilisés étaient le valproate de sodium (58,8%) et le phénobarbital (17,6). Le niveau d’instruction était bas avec 40% de non-scolarisés et 55,6% au primaire. Seuls 6,7% étaient autonomes et 20 patients nécessitaient une aide quasi-constante. Conclusion La prévalence de l’épilepsie chez les patients atteints de TSA varie suivant les études. Une prise en charge globale et multidisciplinaire de l’épilepsie et des TSA améliore les troubles de comportement. Mots clés : Autisme, Epilepsie, pédopsychiatrie, Sénégal. ABSTRACT Description Autism spectrum disorders (ASD) and epilepsy can coexist in the same person, which are factors of bilateral poor prognosis. Purpose To describe the socio-demographic profile of ASD patients and to study the clinical, paraclinical and evolutive aspects of epilepsy among them. Patients and methods We did a cross-sectional and descriptive study in the department of child psychiatry of Fann university hospital in Dakar. We analyzed patients’ files followed between January 2004 and September 2018 for autism’s spectrum disorders with or without epilepsy. We used a standardized survey with several items. Results Forty-five patients with ASD were collected, with an epileptic frequency of 37.8%. Epilepsy had started before the age of 5 years in 94% of cases. Seizures were generalized (58.8%), mainly tonico-clonic (80%), or focal (35.3%), with a frequency of 2 seizures per day to 1 seizure per week. The awake and sleep EEG showed abnormalities mostly in frontal area in 60%, and centro-parietal in 26.6%. Brain imaging was normal in 93.9%, and auditory evoked potential (AEP) normal in 87.2%. Management was multidisciplinary for ASD (neuropsychological, psychomotor, speech therapy) and medical, mainly (82.4%) monotherapy for epilepsy. The main molecules were: sodium valproate (58.8%) and phenobarbital (17.6). The level of education was low with 40% of students out of school and 55.6% in primary school. Only 6.7% were self-reliant and 20 patients needed almost constant assistance. Conclusion The prevalence of epilepsy in patients with ASD varies according to the studies. The global and multidisciplinary management of epilepsy and ASD improves behavioral disorders. Keywords: Autism, child-psychiatry, Epilepsy, Senegal. INTRODUCTION Les troubles du spectre de l’autisme (TSA) correspondent à un groupe de troubles du développement caractérisé par une déficience des interactions sociales, du langage et de la communication, ainsi que des comportements stéréotypés. L’autisme infantile représente environ 33,3% de tous les cas de TSA et est généralement la forme la plus sévère des TSA (4). Les épilepsies sont également un groupe hétérogène de troubles neurologiques relevant d’étiologies multiples, avec des difficultés cognitives et comportementales. L’épilepsie est retrouvée en proportion plus importante sur terrain de troubles du spectre autistique (TSA) (9). La prévalence de l’épilepsie chez les personnes atteintes de TSA a été estimée à 8,9% en l’absence de déficience intellectuelle et à 23,7% chez les personnes ayant une déficience intellectuelle (29). Les enfants déjà diagnostiqués avec un TSA ont un risque élevé de développement ultérieur d’une épilepsie, en particulier en présence d’une déficience intellectuelle (26,29). Environ 30% des personnes atteintes de TSA développent une épilepsie à l’âge adulte, et environ 30% des personnes atteintes d’épilepsie répondent aux critères diagnostiques d’un TSA. L’objectif de notre étude était d’étudier l’incidence et les caractéristiques électrocliniques et évolutives de l’épilepsie chez des patients suivis pour troubles du spectre de l’autisme dans le service de pédopsychiatrie du Centre Hospitalier National Universitaire (CHNU) de FANN à Dakar (Sénégal). Aussi, de manière plus spécifique, d’évaluer la fréquence de l’épilepsie chez les patients atteints de TSA, les syndromes épileptiques associés aux TSA, le retentissement de la maladie sur la scolarité et l’autonomie des patients. PATIENTS ET METHODES : Il s’agissait d’une étude transversale et descriptive de patients suivis entre Janvier 2004 et Septembre 2018 au service de pédopsychiatrie du Centre Hospitalier National de Fann de Dakar (Sénégal). Ont été inclus tous les patients avec un diagnostic confirmé de troubles du spectre de l’autisme, qui sont âgés de moins de 18 ans, sans distinction de sexe ou de race, suivis à l’hôpital de jour dudit service. Ont été exclus de l’étude les patients diagnostiqués avec TSA aux dossiers inexploitables ou incomplets (perdus de vue). La collecte des données était faite grâce à une fiche standardisée de recueil de données comprenant plusieurs items: les facteurs sociodémographiques, antécédents personnels et familiaux, présence ou non d’épilepsie, type / fréquence / âge de début et horaire des crises épileptiques, les résultats de l’électroencéphalogramme de veille et de sommeil, l’imagerie cérébrale, les potentiels évoqués auditifs (PEA), les syndromes électrocliniques retenus, la chronologie de l’épilepsie sur les TSA, la prise en charge de l’épilepsie et des TSA, l’évolution. Les niveaux socio-économiques des parents étaient estimés sur la moyenne des revenus mensuels selon leur profession. L’exploration EEG a été faite ou refaite chez tous les patients épileptiques identifiés dans le Département de Neurophysiologie clinique du CHUN de Fann. RESULTATS Quarante-cinq patients ont été colligés entre Janvier 2004 et Septembre 2018. Le sex-ratio était de 2/1. La majorité des patients (64,4%) était âgée de 6-10 ans (cf tableau I). Quarante-trois patients étaient de nationalité sénégalaise. Vingt patients (44,4%) étaient d’origine semi-urbaine (banlieue), contre 37,7% d’origine urbaine et 17,8% de ruraux. Tous nos patients vivaient avec leurs propres familles. Le niveau socioéconomique était moyen chez la majorité de nos patients (68,9%), bas chez 11,1% et élevé chez 20% d’entre eux. Les parents de 22 patients soient 48,9% avaient un niveau d’instruction universitaire, au contraire de nos patients avec 18 (40%) qui n’étaient pas scolarisés, 25 (55,6%) inscrits à l’école primaire, et 2 (4,4%) au collège (Tableau I). Dans les antécédents, les grossesses s’étaient normalement déroulées chez 34 patients (75,5%). Les complications rencontrées étaient soit une rupture prématurée des membranes de 4 jours chez 1 patient, un accouchement prématuré chez 2 patients, respectivement à 30 et à 32 semaines d’aménorrhée, une souffrance fœtale aigue (SFA) chez 11 patients (24,4%), nécessitant une réanimation, puis suivie d’une hospitalisation au service de néonatologie pour 2 patients. Un patient avait eu une méningite à l’âge de 7 mois et deux autres avaient eu un traumatisme crânien. Une consanguinité parentale était retrouvée chez 10 patients (22,2%) dont 8 de premier degré. Des antécédents familiaux d’épilepsie étaient rapportés chez 5 patients (11,1%), des TSA dans la fratrie chez 4 patients (8,9%) et un retard psychomoteur chez les parents de 2 patients (4,4%) (1 cousin maternel et le père et grand-père paternel). Des antécédents de schizophrénie étaient aussi rapportés chez les parents de 3 patients (6,7%). (Figure 1) Au plan clinique, chez 30 patients (66,7%), les symptômes de TSA étaient constatés avant l’âge de 3 ans, contre 33,3% après l’âge de 3 ans. Vingt-six patients (57,8%), avaient consulté moins de 6 mois après le début des symptômes et quatre (8,9%) dans un délai de 6 à 12 mois, et quinze patients (33,3%) dans un délai de plus d’une année. Tous nos patients avaient bénéficié d’un EEG qui était normal chez 28 patients (62,2%). Trente-trois patients (73,3%) avaient eu une imagerie cérébrale : 21 (63,6%) une tomodensitométrie (TDM) cérébrale et 12 autres (36,4%) une imagerie par résonnance magnétique (IRM) cérébrale. Trente et un patients (93,9%) avaient une imagerie cérébrale normale. Chez deux patients, étaient retrouvées une atrophie cortico-sous-corticale frontale bilatérale et une hypodensité frontale droite en rapport avec des séquelles de méningite. Trente-neuf (86,7%) patients avaient effectué un potentiel évoqué auditif (PEA) dont 34 cas (87,2%) étaient normaux et pathologiques chez 5 autres (12,8%) avec une atteinte rétro-cochléaire bilatérale, une atteinte endocochléaire bilatérale, une hypoacousie mixte, une neuropathie axonale et une surdité isolée. Tous les patients bénéficiaient, en hôpital de jour, d’une prise en charge neuropsychologique, orthophonique et psychomotrice, associée chez 10 patients (22,2%) à une prise charge spécialisée à domicile. L’épilepsie concernait dix-sept patients atteints de TSA (37,8%). Elle avait débuté avant l’âge de 5 ans chez 16 patients (94%) et à l’âge de 9 ans chez un patient. Les TSA étaient apparus avant l’épilepsie chez 7 patients (41,2%) et après l’épilepsie chez 6 patients (35,3%). Les diagnostics de TSA et d’épilepsie ont été faits en même temps chez 4 patients (23,5%). Les crises étaient généralisées chez 10 patients (58,8%), focales chez 6 patients (35,3%) et indéterminées chez 1 patient (5,9%). Les crises généralisées étaient dominées par les crises tonico-cloniques (80%), les crises atoniques (10%) et les absences typiques (10%) étaient plus rares. Les crises focales étaient cloniques (50%) et toniques (50%) avec une bilatéralisation dans 33,3% des cas. Les crises épileptiques avaient une fréquence de 2 crises quotidiennes à une crise hebdomadaire, avec un horaire diurne chez 11 patients, nocturne chez 2 patients et mixte chez 4 patients. Chez les patients avec épilepsie, l’EEG était normal chez 2 patients (11,8%). Des anomalies paroxystiques étaient trouvées chez 15 épileptiques (88,2%). Tous les patients qui avaient des anomalies à l’EEG (17 patients), avaient des anomalies électriques à la veille. Le rythme de fond était normal chez 16 patients et un patient non épileptique avait un ralentissement du rythme de fond. Les anomalies étaient de siège frontal chez 9 patients (60%), centro-pariétal chez 4 patients (26,6%), temporal chez 1 patient (6,7%) et centro-temporal chez 1 patient (6,7%) (Figure 2). Les 2 patients non épileptiques avaient des anomalies frontales à l’EEG de veille. Pour l’EEG de sommeil, le rythme de fond était normal chez 16 patients et ralenti chez un patient non épileptique (le même qui avait un ralentissement au rythme de fond à la veille). Chez les 15 épileptiques, le siège des anomalies paroxystiques était superposable à celui de l’EEG de veille. Au plan syndromique, deux cas d’épilepsie à pointes centro-temporales (EPCT) et un autre de syndrome de pointes ondes continues du sommeil (POCS) ont été identifiés. Pour le reste, le diagnostic syndromique n’a pu être précisé. Quatorze patients soit 82,4% étaient sous monothérapie et les autres sous polythérapie (17,6%). Le valproate de sodium (VPA) était la molécule la plus prescrite chez 10 patients soit 58,8% des cas, suivie du phénobarbital (PB) chez 3 patients (17,6%), de la carbamazépine chez un patient (5,9%) et l’association VPA avec PB chez 3 patients (17,6%). Au plan évolutif, seuls 3 patients (6,7%) étaient autonomes. Vingt-deux patients (48,9%) étaient assez autonomes nécessitant rarement l’aide d’une tierce personne et 20 patients étaient dépendants nécessitant une aide quasi-constante. DISCUSSION De rares séries ont été réalisées sur l’épilepsie chez les personnes atteintes de TSA principalement en Europe et en Amérique du nord. Il s’agissait le plus souvent d’enquêtes rétrospectives (1,27,29). A notre connaissance aucune étude n’a été faite en Afrique et particulièrement au Sénégal sur ce sujet. Les comportements autistiques apparaissaient chez l’enfant souvent avant l’âge de 3 ans (8,12) chez certains auteurs, comme dans notre série avec 30 patients concernés, soit 66,7%. Une prédominance masculine comme dans la littérature (8,22) a aussi été constatée dans notre série. Une association avec l’épilepsie était retrouvée chez des patients autistiques, entre 10 et 12,5% selon les études de Viscidi EW et al. et Ewen JB et al. (24,27). L’étiologie de l’autisme reste encore mal connue, la prédisposition génétique a été prouvée (8, 16, 21) ; cependant d’autres facteurs comme les complications gynéco-obstétricales ont été incriminées. Dans notre étude, l’accouchement prématuré était retrouvé chez 2 patients et une souffrance fœtale aigue rapportée chez 11 patients (24,4%) nécessitant une réanimation. Une notion d’hérédité était retrouvée chez 14 patients soit 31,1% avec des antécédents familiaux d’épilepsie chez 11,1% patients, de TSA dans la fratrie chez 4 patients (8,9%) et psychiatriques parentaux chez 3 patients. Christensen et al. (7) ont rapporté que le risque global de l’épilepsie chez les enfants plus jeunes augmente de 70% si un frère aîné avait un TSA, et augmente de 54% si un frère ou une sœur plus âgée avaient une épilepsie. La fréquence de l’épilepsie chez les enfants atteints de TSA dans notre population d’étude était de 37,8%. Ce résultat est similaire à ceux décrits dans la littérature (1,25,29). La quasi-totalité des patients (94%) avait un âge de début des crises épileptiques précoce avant 5 ans. Viscidi et al. (27) ont rapporté que la prévalence de l’épilepsie chez les enfants atteints de TSA âgés entre 2 et 17 ans est de 12,5%, alors qu’elle est de 26% chez ceux qui ont plus de 13 ans. Des anomalies paroxystiques à l’EEG ont été trouvées chez 37,8% des patients. Hughes et al. (15) ont rapporté que jusqu’à 60% des enregistrements EEG des enfants avec TSA ont des pointes intercritiques. À l’EEG de veille et de sommeil, les anomalies paroxystiques étaient frontales (60%) et centro-pariétales (26,6%), plus rarement temporales (6,7%) et centro-temporales (6,7%). Il en était de même dans l’étude de Donies Masmoudi et al. (19). Matsuo et al. ont rapporté des anomalies frontales dans 76% des cas, centro-pariétales dans 15% des cas, occipitales dans 6% des cas et temporales chez 2% des patients (20). Sur le plan radiologique, le rôle de l’imagerie par tenseur de diffusion (ITD) est primordial en mettant en évidence les anomalies de la substance blanche pendant le développement précoce des TSA et de l’épilepsie (3). Cependant cet examen paraclinique n’était pas disponible. L’IRM cérébrale permet de mettre en évidence d’éventuelles anomalies cérébrales. Cependant, compte tenu du niveau socioéconomique faible, 63,6% des patients avaient réalisé une tomodensitométrie cérébrale. Aucun patient ne présentait de malformations du développement cortical, à type de désorganisation cellulaire, d’hétérotopies ou dysplasies souvent décrites dans la littérature (28). Toutefois, l’imagerie encéphalique pouvait être normale, comme retrouvée par Boddaert et al. (5) chez 47% de ses patients ; dans notre étude, elle l’était chez 93,9% des patients. Des anomalies temporales, de la substance blanche et/ou des espaces de Wirshow-Robin ont été décrites (28), et retrouvées chez 3 patients soit 6,1% des cas. Tous les patients ont bénéficié d’une prise en charge neuropsychologique, en psychomotricité et orthophonie en hôpital de jour, associée à une prise charge spécialisée à domicile chez 22,2% des patients. La prise en charge psychosociale se basait sur le profil comportemental, développemental et psychanalytique. La prise en charge médicale concernait les patients épileptiques avec des médicaments antiépileptiques majoritairement en monothérapie, principalement le phénobarbital (82,4%), et du valproate de sodium (58,8%). Plusieurs études rapportent une amélioration des symptômes fondamentaux de l’autisme, de l’impulsivité et de l’agressivité des enfants avec TSA et épilepsie ou avec TSA avec anomalies paroxystiques à l’EEG sans crises épileptiques, traitées par l’acide valproïque (13). La lamotrigine, le levétiracétam et les autres modalités thérapeutiques de l’épilepsie n’ont pu être utilisés car indisponibles dans nos contrées. Malgré les travaux multiples sur ce sujet, la nature et la fréquence de l’association entre les épilepsies et les TSA restent mal compris. Des mécanismes sous-jacents, des facteurs de risque génétiques et environnementaux communs ont été identifiés. Ainsi une étude à long terme s’avère nécessaire à la compréhension des deux troubles. CONCLUSION La variation dans la prévalence de l’épilepsie chez les patients atteints de TSA semble être en rapport direct avec les différences dans les caractéristiques de l’échantillon entre les études. Toutefois, les profils épidémiologiques semblent similaires quelles que soient les populations étudiées. Une particularité semble se profiler dans notre contexte avec des antécédents familiaux plus lourds dont une consanguinité parentale, sous tendus par un environnement périnatal peu favorable à travers l’existence d’une souffrance cérébrale, une méningite et/ou un traumatisme crânien en période néonatale. La prise en charge médicale de l’épilepsie, intégrée dans une prise en charge globale et multidisciplinaire, chez les patients atteints de TSA permet d’améliorer les troubles de comportement inhérents chez ces enfants. Conflit d’intérêts : Aucun des auteurs n’a de conflit d’intérêts à signaler. Divulgation : L’étude n’a reçu aucun soutien financier ni dans sa mise en œuvre, ni pour une orientation des résultats. La seule guidance fut l’intérêt scientifique. Remerciements: Nous tenons à remercier les personnes sans lesquelles cette étude n’aurait pas été possible.
Tableau I : Paramètres socio-démographiques des patients
 Figure 1 : Répartition suivant les antécédents des patients
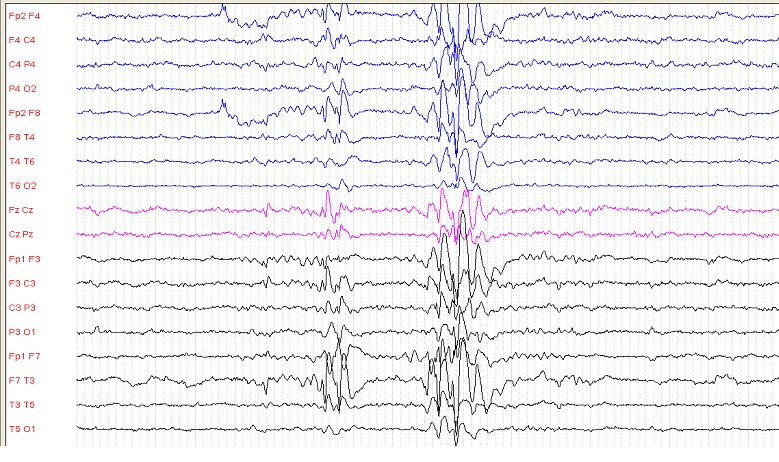 Figure 2 : Illustration EEG de veille d’un garçon de 14 ans.
REFERENCES
|
|||||||||||||||||||||||||||||||||||||||||||
© 2002-2018 African Journal of Neurological Sciences.
All rights reserved. Terms of use.
Tous droits réservés. Termes d'Utilisation.
ISSN: 1992-2647