
|
|
|
CASE REPORT / CAS CLINIQUE
SYNDROME DE LA PERSONNE RAIDE AVEC ANTICORPS ANTI-GAD NEGATIF EN COTE D’IVOIRE : UN CAS CLINIQUE AVEC REVUE DE LA LITTERATURE
STIFF PERSON SYNDROME WITH NEGATIVE AUTOANTIBODIES AGAINST GAD IN IVORY COAST : A CASE REPORT AND LITERATURE REVIEW
E-Mail Contact - BONY Kotchi Elisée :
bonyk2004@yahoo.fr
RESUME Le syndrome de la personne raide (SPR) est un trouble auto-immun rare du système nerveux central caractérisé par une raideur progressive des membres et des spasmes musculaires. Ce syndrome a été très peu rapporté dans la littérature en Afrique sub-saharienne. Nous présentons l’observation du premier cas de syndrome de la personne raide (SPR) diagnostiqué en Côte d’ivoire chez un patient de 20 ans, reçu en consultation pour une rigidité généralisée d’installation progressive. Cette rigidité qui avait débuté aux membres inférieurs était associée à des spasmes distaux accentués par les émotions et les stimulations tactiles. Ses antécédents étaient sans particularité. L’examen neurologique a objectivé la rigidité des membres inférieurs isolée sans troubles des réflexes. L’IRM médullaire et L’IRM cérébrale étaient normales. L’ENMG a objectivé une activité continue de potentiels d’unités motrices sur le tracé de détection spontanée au niveau de plusieurs muscles des membres inférieurs (quadriceps, jambiers antérieurs). La sémiologique clinique et les résultats de l’EMG ont permis de retenir le diagnostic de SPR. Le dosage des Ac anti-GAD dans le sang était négatif. L’évolution clinique a été satisfaisante au bout de deux semaines sous baclofène et des séances de rééducation fonctionnelle. Mots clés : syndrome de la personne raide ; stiff-leg syndrome ; Afrique ; Côte d’ivoire SUMMARY The stiff person syndrome (SPS) is a rare autoimmune disorder of the central nervous system characterized by progressive limb stiffness and muscle spasms. Few cases of SPS are described in the literature in sub-Saharan Africa. We herein report the first case of SPS diagnosed in Ivory Coast (West Africa), in a 20-years-old man, admitted with the complaint of gait disturbance, rigidity and muscle spasms of lower limbs of progressive installation. His rigidity began from the lower limbs and was associated with distal extremities spasms accentuated by emotions and tactile stimulations. His had no medical history. The neurological examination showed stiffness of the lower limbs without reflexes disturbance. Medullary MRI and brain MRI were normal. The electromyography (ENMG) recorded with needle electrodes showed spontaneous and continuous motor unit discharges in lower extremities. Clinical semiology and ENMG analysis allowed to retain SPS as diagnosis. The Serum autoantibody panel for anti-GAD was negative. The clinical course was good after two weeks of treatment with baclofen and functional rehabilitation sessions. Key words: stiff person syndrome; stiff-leg syndrome; Africa; Ivory Coast INTRODUCTION Le syndrome de la personne raide (SPR) est un trouble rare du système nerveux central caractérisé par une raideur progressive des membres et des spasmes musculaires. Le SPS anciennement dénommé syndrome de l’homme raide, est une maladie auto-immune caractérisée par la présence dans 60-80% des cas d’autoanticorps dirigés contre l’acide glutamique décarboxylase (autoanticorps anti-GAD). En Afrique subsaharienne, la littérature rapporte très peu de cas chez le noir africain (2). Nous rapportons un cas clinique de syndrome de la personne raide avec anticorps antiGAD négatif diagnostiqué en Côte d’ivoire chez un patient de 20 ans. OBSERVATION Un patient âgé de 20 ans, a été vu en consultation de Neurologie en Mars 2018 pour une rigidité généralisée, axiale et des membres, associée à des spasmes distaux évoluant par accès. Le début des symptômes qui se situe en 2014 a été progressif, sans contexte infectieux, d’abord aux membres inférieurs, d’aggravation régulière, avec des chutes fréquentes au moment des paroxysmes. Ces spasmes aux membres inférieurs étaient à l’origine d’une gêne à la marche, occasionnant l’utilisation d’une canne. Cette rigidité était accentuée par les émotions et les stimulations tactiles et disparaissait pendant le sommeil. De plus, Il a rapporté une notion de dysphagie aux liquides et une sensation de laryngopharyngospasme. Les antécédents médicaux personnels et familiaux étaient sans particularité marqués notamment par l’absence de diabète, de prise toxique, de comitialité, de traumatisme et d’affection hématologique. L’examen général était normal. L’examen neurologique a mis en évidence une rigidité des membres inférieurs, une absence de déficit sensitivo-moteur. Les réflexes ostéo tendineux étaient normaux de même que les fonctions génito-vésico-anales. En outre, on notait une légère accentuation de la lordose lombaire et une absence de myoclonie et de trouble cognitif. L’IRM médullaire et l’IRM cérébrale étaient normales. L’ENMG a mis en évidence une activité musculaire quasi continue au niveau des membres inférieurs compatible avec un syndrome de la personne raide. L’échographie thyroïdienne et le scanner thoracique étaient normaux. L’examen du liquide cérébro-spinal n’a pu être réalisé en raison du refus de la ponction lombaire par le patient. Le bilan biologique sanguin standard, comportant l’hémogramme, la VS; la C Réactive Protéine, , la créatininémie, la glycémie veineuse à jeun, les transaminases, .était sans particularité. Le taux de TSH était normal. La recherche d’autoanticorps anti GAD dans le sang était négative. Les sérologies virales du HTLV1, du VIH et de l’Hépatite B étaient négatives. Le facteur rhumatoïde, les Ac antithyroïdiens, les Ac anti Récepteurs de l’acétylcholine n’ont pas été recherchés. Un traitement symptomatique à base de baclofène par voie orale à la dose de 20 mg par jour pendant 2 semaines associé à des séances de rééducation fonctionnelle a entraîné une régression totale des signes fonctionnels et physiques. DISCUSSION La première description d’un syndrome de la personne raide (SPR) a été faite par Moersch et Woltman en 1956 dans une série de 13 personnes (4). Le SPR est une maladie rare dont l’incidence est faible, entre 0.5 et 2 pour 1 000 000 d’habitants (2). En Afrique noire, la seule série sur le SPR a été rapportée par Decker Marieke et al en Tanzanie chez 9 personnes (2). Nous rapportons le premier cas diagnostiqué en Afrique de l’Ouest en Côte d’Ivoire. L’âge moyen de début du SPR est situé entre les quatrièmes et les cinquièmes décades, mais des cas de SPR ont été rapportés chez des sujets plus jeunes comme notre patient (3). Dans les formes typiques, les femmes sont fréquemment plus touchées que les hommes, ce qui explique le remplacement de la terminologie « stiff-man syndrome » par « stiff-person syndrome » (3). Le mécanisme de survenue du SPR serait une dérégulation de l’inhibition GABAergique conduisant à une contraction musculaire soutenue, une raideur et une rigidité axiale et des membres. Les détails du mécanisme pathogénique exact restent à élucider. Il existe cependant des preuves d’un processus auto-immun sous-jacent impliquant potentiellement à la fois les lymphocytes B et T. Le processus auto-immun est également sous-tendu par la mise en évidence dans le sérum des patients d’autoanticorps comme les anticorps anti-glutamique décarboxylase (anti-GAD) et / ou anti-amphiphysine (3). Au plan clinique, le cas rapporté est similaire à ceux décrits dans la littérature caractérisés par un début insidieux avec une évolution au-delà d’un an (2 ; 3), la raideur et les spasmes musculaires déclenchés par des stimuli. Dans notre observation il s’agissait d’émotions et de stimuli tactiles. La présence des troubles initialement localisés aux membres inférieurs correspond à l’une des formes cliniques du SPR : le « stiff-leg syndrome ». La marche spastique de notre patient nous a fait évoquer une paralysie spastique tropicale (PST) progressive dans le cadre des neuromyélopathies à HTLV-1 (1). La normalité de L’IRM médullaire, et de la sérologie du HTLV-1 ont permis de récuser le diagnostic de myélopathie à HTLV-1. L’ENMG en montrant une activité musculaire quasi continue des membres inférieurs, a orienté vers le diagnostic d’un « stiff-leg syndrome ». En effet, notre patient avait les 7 critères diagnostiques du SPR. Il s’agit de l’apparition d’une raideur des muscles axiaux, de l’évolution lente de la rigidité qui gagne les muscles des racines des membres entraînant une difficulté lors de la réalisation des mouvements volontaires de la marche, de la déformation fixe du rachis, des spasmes paroxystiques provoqués par un mouvement, un bruit brusque ou une émotion , d’un examen sensitivomoteur normal, de l’absence de trouble cognitif et d’un ENMG caractérisé par une activité continue de potentiels d’unités motrices sans signe de dénervation, qui disparait sous diazépam en intraveineux ou per os. (3). Devant une rigidité généralisée progressive un syndrome parkinsonien est facilement éliminé par l’analyse sémiologique qui objective une akinésie, un tremblement de repos et une hypertonie extrapyramidale. Une atteinte médullaire est éliminée par les anomalies de l’IRM médullaire, certaines myopathies avec les signes musculaires permanents et les anomalies biologiques (enzymes musculaires, biopsie musculaire). Le diagnostic différentiel est plus difficile avec la neuromyotonie acquise ou syndrome d’Isaac. Cette myotonie d’origine axonale motoneuronale, persiste pendant le sommeil, à la différence de la rigidité du SPR qui est d’origine centrale et qui disparait pendant le sommeil (3). Le dosage sanguin à postériori des Ac anti-GAD ainsi que les sérologies virales (HTLV1, VIH, Hépatite B) étaient négatifs. La recherche d’autres affections auto-immunes par le dosage du Facteur rhumatoïde, des Ac antithyroïdiens, des Ac anti Récepteurs de l’acétylcholine n’a pas été possible. Mais dans les formes atypiques de SPR, on a décrit une positivité moins fréquente (15 à 20%) des Ac anti-GAD et une moindre association aux autres maladies auto-immunes (3). La présence d’Ac anti-îlots de Langerhans, d’Ac antithyroperoxydase, d’Ac antithyroglobuline et d’Ac anti-amphiphysine est également rapportée (3). Le traitement du SPR comporte deux volets : un traitement symptomatique et un traitement étiopathogénique. Le traitement symptomatique qui repose sur les myorelaxants tels que le baclofène, ou le diazepam a une efficacité très variable sur les spasmes. D’autres molécules telles que le vigabatrin, la gabapentine sont également efficaces sur les spasmes. (3) Le traitement étiopathogénique comporte la corticothérapie, qui pour certaines équipes est un traitement de choix (3), de même que les immunoglobulines intraveineuses et la plasmaphérèse pour leur efficacité sur la rigidité. Le rituximab qui pourrait être une alternative prometteuse n’a, à l’heure actuelle, pas encore fait la preuve de son efficacité. Son utilisation chez des patients séronégatifs aux anticorps anti-GAD n’a pas été rapportée (5). L’évolution de notre patient a été rapidement satisfaisante sous baclofène marquée par la disparition totale des spasmes et de la rigidité. CONCLUSION Le premier cas de SPR dans sa forme stiff-leg syndrome avec Ac anti-GAD négatif est rapporté en Côte d’Ivoire. Le diagnostic a été évoqué sur la conjonction de critères cliniques et ENMG. Le traitement uniquement symptomatique par du bacloféne a entrainé une évolution rapidement favorable. Conflit d’intérêt : Les auteurs ne déclarent aucun conflit d’intérêt. Illustration : STIFF PERSON SYNDROME WITH NEGATIVE AUTOANTIBODIES AGAINST GAD IN IVORY COAST : A CASE REPORT AND LITERATURE REVIEW. 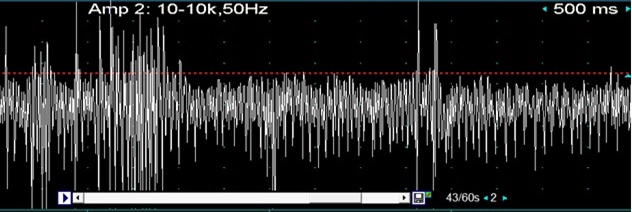 Figure 1 : Electromyographie : tracé de détection spontanée au niveau du jambier antérieur gauche montrant une activité continue de potentiels d’unités motrices REFERENCES
|
© 2002-2018 African Journal of Neurological Sciences.
All rights reserved. Terms of use.
Tous droits réservés. Termes d'Utilisation.
ISSN: 1992-2647