
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
CLINICAL STUDIES / ETUDES CLINIQUES
SYNDROME DE WEST : PROFIL DES PATIENTS AU CHU DE FANN DAKAR-SENEGAL
WEST SYNDROME: PATIENT PROFILE AT FANN DAKAR-SENEGAL TEACHING HOSPITAL
E-Mail Contact - BOUDZOUMOU Diagambana Bertandrie Estelle :
dr.estelle2014@gmail.com
RESUME Introduction Le syndrome de West ou encéphalopathie épileptique avec hypsarythmie est fréquente chez le nourrisson au Sénégal. Son pronostic est étroitement lié à la pathologie sous-jacente .Il est principalement secondaire à des causes périnatales. Patients et méthodes Il s’est agi d’une étude rétrospective réalisée à la Clinique Neurologique du CHU de Fann et à l’Hôpital national pédiatrique Albert Royer, à Dakar-Sénégal, d’Avril 2012 à Aout 2018. Sur les dossiers médicaux et les registres de suivi, nous avons recueilli les caractéristiques socio-démographiques, les données cliniques, les données paracliniques, les modalités thérapeutiques, et l’évolution des patients. Résultats Nous avons colligé 37 nourrissons avec une prédominance masculine (59%), dont l’âge moyen au moment du diagnostic était de 8 ± 4mois et l’âge moyen d’apparition des spasmes était de 3mois. L’EEG montrait une hypsarythmie chez tous les patients. Les causes étaient dominées par l’atrophie cortico- sous corticale (21,62%). En première intention, le traitement associait souvent le valproate de sodium et des corticoïdes (35,13%). La pharmacorésitance nécessitait d’associer valproate de sodium au vigabatrin (8%). L’évolution clinique était surtout marquée par un arrêt des crises (24,32%), et une persistance des crises (19%). Discussion et Conclusion Le syndrome de West est une encéphalopathie épileptique du nourrisson dominé par des spasmes en flexion. Dans notre contexte, les facteurs de risque sont l’absence de suivi prénatal, la souffrance néonatale, l’accouchement dystocique. La polythérapie permet d’arrêter les crises, qui dans certains cas peuvent devenir pharmacorésistantes. L’imagerie par résonnance magnétique et le bilan génétique sont presque inaccessibles, de même que le vigabatrin dont le cout est élevé. Mots-clés : encéphalopathie, épilepsie, hypsarythmie, spasmes. Sénégal, Syndrome de WEST ABSTRACT Introduction West syndrome or epileptic encephalopathy with hypsarrhythmia is common in infants in Senegal. Its prognosis is closely related to the underlying pathology. It is mainly secondary to perinatal causes. Patients and methods This was a retrospective study conducted at the Fann University Hospital Neurological Clinic and at the Albert Royer National Pediatric Hospital in Dakar, from April 2012 to August 2018. On medical records and follow-up registers, we collected socio-demographic characteristics, clinical data, paraclinical data, therapeutic modalities, and patient outcomes. Results We collected 37 infants with male predominance (59%), whose mean age at diagnosis was 8 ± 4 months and the mean age at the onset of spasm was 3 months. EEG showed hypsarrhythmia in all patients. The causes were dominated by cortico-subcortical atrophy (21.62%). As a first-line treatment, sodium valproate and corticosteroids were often combined (35.13%). Drug resistance required the combination of valproate sodium and vigabatrin (8%). The clinical course was mainly marked by a cessation of seizures (24.32%), and a persistence of seizures (19%). Discussion and Conclusion West’s syndrome is an infantile epileptic encephalopathy dominated by flexion spasm. In our context, the risk factors are the absence of prenatal follow-up, neonatal distress, and obstructed labor. Combination therapy can stop seizures, which in some cases may become drug-resistant. Magnetic resonance imaging and gene balance are almost inaccessible, as is vigabatrin, which is expensive. Keywords: epilepsy, hypsarrhythmia, Senegal, West syndrome, INTRODUCTION L’épilepsie est un dysfonctionnement cérébral caractérisé par une prédisposition chronique à la génération de crises épileptiques et par les conséquences neurobiologiques, cognitives, psychologiques et sociales de cet état. La définition de l’épilepsie nécessite la survenue d’au moins une crise épileptique. Une crise épileptique est la survenue transitoire de signes et symptômes liés à une activité neuronale anormale, excessive ou synchrone, dans le cerveau. Un syndrome épileptique définit une forme d’épilepsie caractéristique par l’âge de survenue des crises, son évolution dans le temps, le type de crise et surtout par le motif électroencéphalographique. Les données de l’électroencéphalogramme (EEG) sont capitales dans cette classification. Le syndrome de West ou encéphalopathie infantile avec hypsarythmie est une forme rare d’épilepsie. Il débute le plus souvent avant un an chez un nourrisson avec un retard de développement psychomoteur préalable ou chez un nourrisson normal avec un arrêt du développement psychomoteur, puis régression [16]. Le pronostic est étroitement lié à la pathologie sous-jacente et à la précocité du traitement [5]. Au Sénégal, le syndrome de West demeure l’encéphalopathie épileptique la plus fréquente du nourrisson. Il est principalement secondaire à des causes périnatales, favorisées par un mauvais suivi prénatal .C’est ainsi que nous avons décidé de réaliser cette étude dont l’objectif principal était de décrire les caractéristiques du syndrome de West. PATIENTS ET METHODES Il s’est agi d’une étude rétrospective et descriptive, qui s’est déroulée à la clinique neurologique du Centre Hospitalier Universitaire de Fann, et à l’Hôpital national pédiatrique Albert Royer, à Dakar-Sénégal. Nous avons inclus les enfants suivis pour un syndrome de West entre Avril 2012 et Aout 2018. Les enfants souffrant d’une encéphalopathie épileptique sans hypsarythmie et ceux ayant des dossiers incomplets, étaient exclus de l’étude. A l’aide de dossiers médicaux, nous avons recueilli les données biographiques (âge, sexe), les données cliniques relatives au syndrome de West, les données électro-encéphalographiques, les données radiologiques (Tomodensitométrie cérébrale, Imagerie par résonnance magnétique), les données thérapeutiques et évolutives. Les différents paramètres étudiés étaient : l’âge au moment du diagnostic, l’âge d’apparition des spasmes, la nature des spasmes, les autres types de crises épileptiques, le développement psychomoteur, le pattern électro-encéphalographique, les modalités thérapeutiques et évolutives. Les données ont été saisies sur Microsoft Word. Nous avons procédé aux calculs de fréquence, de la moyenne, de la médiane, de l’écart-type. L’analyse des données a été faite sur Microsoft Excel 2010. RESULTATS Nous avons colligé dans cette étude 37 nourrissons, dont 15 filles (41%) et 22 garçons (59%), avec un sex ratio à 1,47. Les âges extrêmes étaient de 3 et 24 mois. La moyenne d’âge était de 8,8 mois ± 4,04 mois, avec une médiane de huit mois. L’âge moyen d’apparition des spasmes était de 3,1 mois avec des extrêmes de 2 et 7 mois. Le suivi prénatal avait été réalisé chez une seule femme en gestation. La grossesse a été menée à terme chez 15 enfants (40,54%), et un enfant était issu d’une grossesse prématurée (2,70%). Dix-sept enfants (45,95%) étaient nés d’un accouchement eutocique, et six (16,21%) d’un accouchement dystocique. Nous avons noté des antécédents de souffrance néonatale chez sept nourrissons (18,91%). Un nourrisson (2,70%) avait des antécédents de trisomie 21. Les spasmes épileptiques étaient présents chez tous les nourrissons. Ils apparaissaient entre un et sept mois de vie. Ces spasmes étaient en flexion chez 26 enfants (72,97%), en flexion et extension chez deux nourrissons (5,40%). Leurs caractéristiques n’étaient pas précisées chez les autres nourrissons. Les autres types de crises sont représentés dans le tableau I. Un patient avait présenté des convulsions néonatales avant le début des spasmes. Le développement psychomoteur était altéré chez 29 patients (78,37%). Le contact social était pauvre chez 17 nourrissons (45,95%). Il était bon chez quatre enfants (10,81%). La microcéphalie était retrouvée chez deux nourrissons (5,40%). Nous avons représenté les autres signes neurologiques associés dans le tableau II. L’électroencéphalogramme était réalisé chez tous les patients, et montrait une hypsarythmie (figures 1 et 2a) chez tous les nourrissons (100%). Les signes radiologiques étaient étudiés au scanner cérébral (tableau III), et/ou à l’Imagerie par résonnance magnétique qui était normale chez trois nourrissons (8,11%). Elle montrait une hydrocéphalie chez un nourrisson (2,70%) et une encéphalomalacie post-anoxique chez un nourrisson (2,70%). Les molécules utilisées pour le traitement étaient : le phénobarbital, le valproate de sodium, la carbamazépine, le clonazépam, le diazépam, le piracétam, la bétaméthasone et la prednisone. La corticothérapie était de courte durée associée au traitement adjuvant à base de calcium. En première intention, (Tableau IV), les nourrissons étaient traités à base d’une monothérapie, une bithérapie, ou une trithérapie. La kinésithérapie était associée au traitement médicamenteux. L’évolution clinique sous traitement était variable (figure 3). Les acquisitions psychomotrices s’étaient améliorées chez quatre enfants (10,81%). L’électroencéphalogramme était devenu quasi normal (figure 2b) chez deux nourrissons (5,40%). DISCUSSION Notre étude révélait une prédominance masculine. Nos données sur la prédominance masculine, l’âge au moment du diagnostic (3 à 24 mois) et l’âge d’apparition des spasmes (2 à 7 mois) sont globalement comparables à ceux retrouvés dans la littérature. Au Canada, en 2001, Brna et al. avaient noté une prédominance masculine à 60%. En 2017, au Kosovo, Zeka et al. [17] avaient également retrouvé une prédominance masculine, avec une moyenne d’âge d’apparition des spasmes de 2,5 mois. Au Portugal, Matta et al. [13], en 2007, avaient noté une prédominance masculine à 62%, avec une moyenne d’âge au moment du diagnostic de 4,9 mois. En Tunisie, Dhahri et al. [5] avaient retrouvé une moyenne d’âge au moment du diagnostic de 8,1 mois. En Afrique du Sud, Keshave et al. [12] en 2017, avaient aussi obtenu une prédominance masculine (87,5%) sur un échantillon de huit patients. Ils rapportaient un âge moyen de début des spasmes de 4,4 mois avec des extrêmes de 1 et 9 mois. Et la moyenne d’âge au moment du diagnostic était de 7,5 mois avec des extrêmes de 3 et 18 mois. Une étude réalisée précédemment au Sénégal en 2017, par Halima et al. [10] montrait une moyenne d’âge d’apparition des spasmes de 6 mois, avec un âge moyen de 10 mois au moment du diagnostic. Le taux de suivi prénatal était bas, d’après nos résultats. En effet, il n’avait été noté que dans un seul cas (2,70%). La consultation prénatale permet de prendre les mesures appropriées pour que l’accouchement se déroule au bon moment (programmer si nécessaire), au bon endroit (référer s’il le faut), et dans les meilleures conditions (considérer les particularités de chaque parturiente) [6]. Les consultations prénatales au troisième trimestre seraient l’occasion de faire un pronostic de l’accouchement grâce à l’utilisation du score de risque de dystocie [14]. Le taux de naissances par accouchement eutocique était élevé. Ce taux de naissance élevé par accouchement eutocique était aussi observé en Afrique du sud par Keshave et al. [12]. Les facteurs de risque retrouvés dans notre série étaient par ordre croissant, l’absence de suivi prénatal, la souffrance néonatale, l’accouchement dystocique et la prématurité. Dans notre étude, tous les patients présentaient des spasmes avec une prédominance des spasmes en flexion. Le syndrome de West est une encéphalopathie épileptique qui se traduit par des spasmes en flexion plus fréquents que les spasmes en extension et les spasmes mixtes. Les spasmes mixtes associent la flexion des membres supérieurs et l’extension des membres inférieurs [16]. Les spasmes en flexion ont la même valeur sémiologique que les spasmes en extension. Plus de la moitié des patients avaient un développement psychomoteur altéré. Le contact social était pauvre dans la moitié de notre échantillon. Keshave et al. [12] avaient observé des troubles du développement psychomoteur chez tous les enfants (100%). Zeka et al. [17] avaient aussi noté des troubles du développement psychomoteur dans 13 cas (78,6%). L’électroencéphalogramme était anormal, comportant une hypsarythmie chez tous les nourrissons. Keshave et al. [12], avaient également observé une hypsarythmie sur l’électroencéphalogramme chez tous les patients. Dhahri et al. [5], notaient une hypsarythmie typique sur l’électroencéphalogramme chez 21 enfants dans un échantillon de 30 cas. Dans notre série, les causes étaient aussi variées, avec une prédominance de l’atrophie cortico-sous-corticale (21,62%), suivie de l’atrophie fronto-pariétale (10,81%), des séquelles d’ischémie cérébrale (10,81%) et des malformations cérébrales. Les autres causes les moins fréquentes étaient l’hémorragie cérébrale, l’encéphalomalacie post-anoxique. L’analyse génétique à la recherche d’aberration chromosomique n’a pas été réalisée dans notre étude. Dans la série de Keshave et al. [12], les anomalies radiologiques étaient dominées par l’atrophie cortico-corticale et l’agénésie du corps calleux. Dhahri et al. [5] avaient noté des examens neuroradiologiques pathologiques chez 22 cas et deux enfants avaient une aberration chromosomique, sur un échantillon de 30 cas. Selon la littérature [4], les étiologies du syndrome de West sont très variées, identifiées dans plus de 50% des cas. Les causes les plus fréquentes, par ordre décroissant, sont l’encéphalopathie hypoxique-ischémique (10%), les anomalies chromosomiques (8%), les malformations (8%), l’accident vasculaire cérébral périnatal (8%), la sclérose tubéreuse de Bourneville (7%) et la leucomalacie périventriculaire, ou l’hémorragie cérébrale (5%). En première intention [tableau IV], les nourrissons recevaient majoritairement une bithérapie à base de valproate de sodium et de corticoïdes (bétaméthasone ou prednisone). En monothérapie, la molécule la plus prescrite était le valproate de sodium, suivie du phénobarbital. La pharmacorésitance nécessitait de faire recours en deuxième intention [tableau V], majoritairement à une bithérapie à base de valproate de sodium et corticoïdes, ou valproate de sodium et vigabatrin. Les corticoïdes administrés étaient la prednisone ou la bétaméthasone. Dans l’échantillon de Keshave et al. [12], tous les huit enfants (100%) avaient reçu du valproate de sodium. La moitié des enfants avait reçu une corticothérapie orale à base de prednisone. Et l’hormone adrénocorticotrophique était administrée chez 3 enfants. Seuls deux enfants avaient reçu du vigabatrin. Dans la série d’Ayadi et al. [1], les patients recevaient une association de plus de deux antiépileptiques en cas de persistance des crises. Dans l’étude de Dhahri et al. [5], le traitement était basé essentiellement sur le vigabatrin et les corticoïdes. Des études récentes suggèrent que l’initiation précoce du traitement, soit avec un traitement hormonal ou le vigabatrin, améliore à long terme les troubles cognitifs. En outre, il a été observé une meilleure efficacité de l’ACTH comparé au vigabatrin dans les troubles cognitifs pour les formes cryptogéniques. Différents corticostéroïdes (hydrocortisone, prednisone, prednisolone ou dexaméthasone), et antiépileptiques peuvent être utilisés en deuxième intention lorsque les médicaments de première ligne sont inefficaces ou contre-indiqués. Environ 25 à 40% des patients dont les spasmes persistent, avec un retard psychomoteur, peuvent être candidats à la chirurgie [4]. La chirurgie n’est pas indiquée si la résection corticale peut créer un nouveau déficit neurologique, en cas de lésions cérébrales diffuses ou de maladie métabolique [9]. Dans notre série, l’évolution clinique [figure 3] était surtout marquée par un arrêt des crises (24,32%) et une persistance des crises chez 18,92%. Et au cours de la surveillance paraclinique, seuls deux de nos patients (5,40%) avaient un électroencéphalogramme de suivi normal. Keshave et al. [12] avaient noté un arrêt des crises dans la moitié de leur échantillon. La réduction de la fréquence des spasmes était aussi retrouvée chez la moitié des patients. Le devenir du syndrome de West est difficilement prévisible, cette encéphalopathie étant un grave syndrome souvent réfractaire au traitement et fréquemment associé à un retard mental [7]. CONCLUSION Le syndrome de West est une encéphalopathie épileptique fréquente chez le nourrisson. Le tableau clinique est dominé par des spasmes en flexion. Les crises associées sont majoritairement représentées par les myoclonies. Les facteurs de risque sont dominés par l’absence de suivi prénatal, la souffrance néonatale, l’accouchement dystocique. La polythérapie permet d’arrêter les crises, mais certains cas peuvent demeurer pharmacorésistants. Dans notre contexte, l’imagerie par résonnance magnétique n’est pas disponible chez la majorité des enfants, de même que le bilan génétique. Le vigabatrin est rarement prescrit à cause de son inaccessibilité et de son coût élevé dans nos régions. 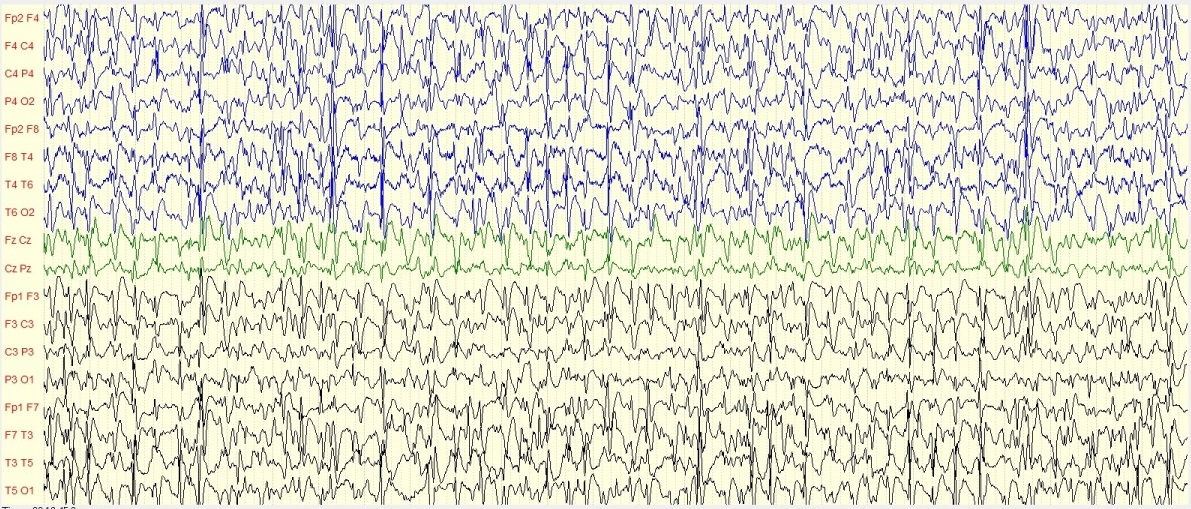 Figure 1 : Tracé EEG de sommeil montrant une hypsarythmie avec des pointes-ondes généralisées chez une fillette de 24 mois
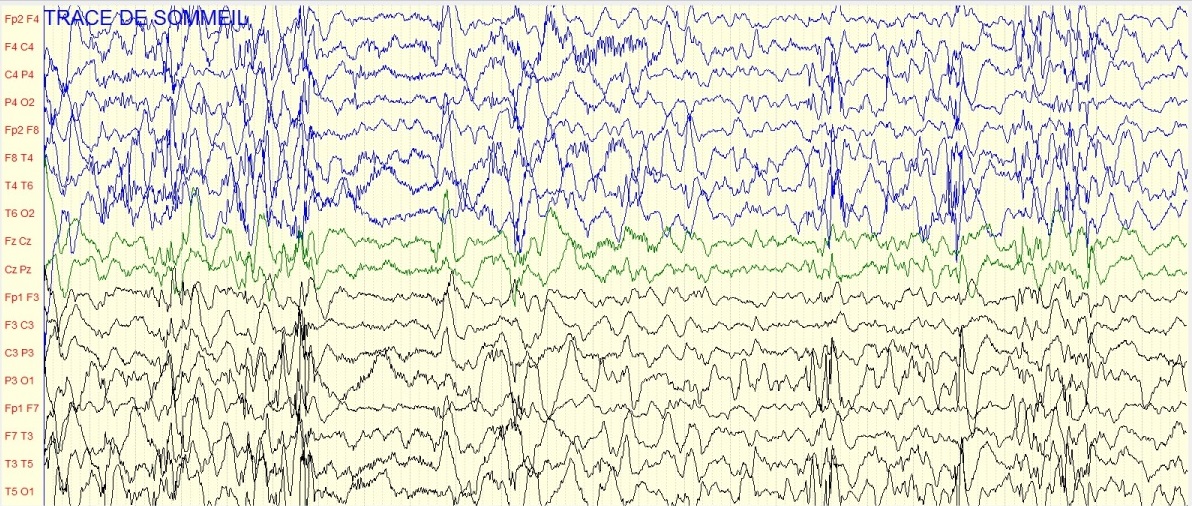 Figure 2a : Tracé EEG de sommeil montrant une hypsarythmie chez un garçon de 11 mois
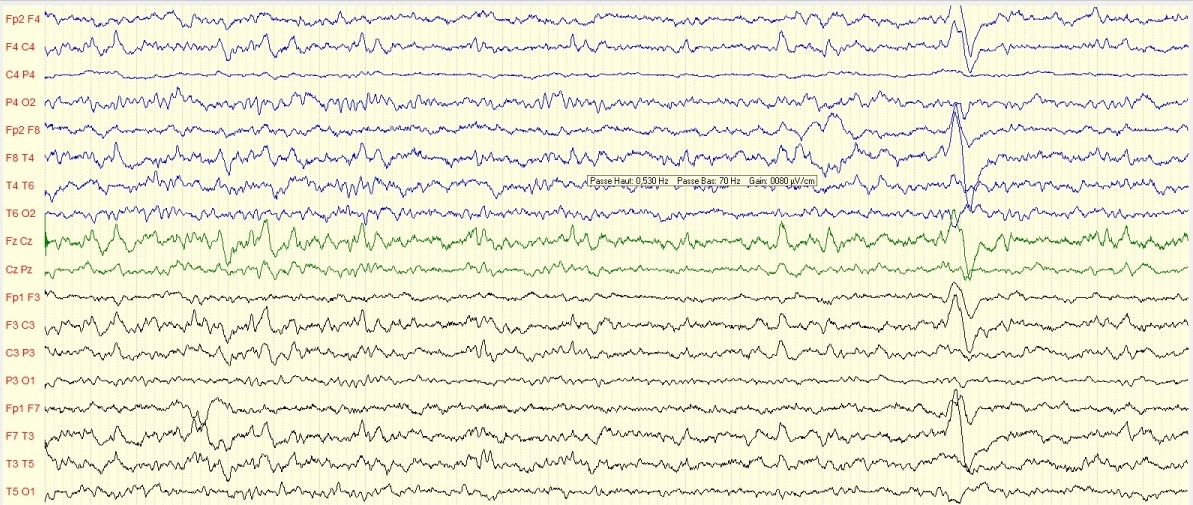 Figure2b : Tracé EEG de contrôle quasi-normal avec disparition de l’hypsarythmie et des ébauches de figures physiologiques du sommeil
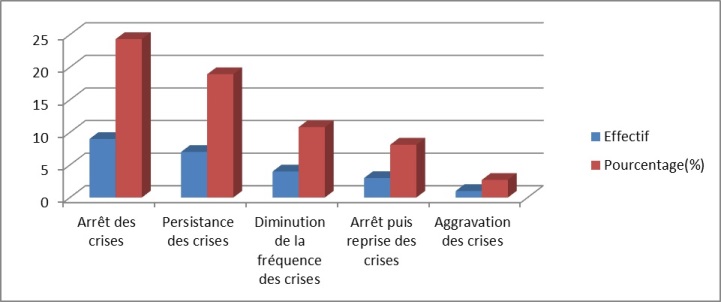 Figure 3 : Aspects évolutifs Tableau I : Autres types de crises
Tableau II : Signes neurologiques associés
Tableau III : Signes radiologiques
Tableau IV : Schéma thérapeutique en première intention
Tableau V : Schéma thérapeutique en deuxième intention
REFERENCES
|
© 2002-2018 African Journal of Neurological Sciences.
All rights reserved. Terms of use.
Tous droits réservés. Termes d'Utilisation.
ISSN: 1992-2647