
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Basic Set of Neurosurgical Instruments : Description of Set The set is based on experience gained under relatively simple conditions where there is no access to highly sophisticated imaging technologies such as CTand MRI nor to a major neurosurgical centre. The quality of the instruments is excellent and guarantees that the set is absolutely sufficient for straightforward basic neurosurgical procedures. Power-driven tools are intentionally not included be working in every environment. Haemostatic agents, silver clips and bone wax are also not part of the set. Basic Set of Neurosurgical Instruments Handling of Order, Payment and Delivery For further information please contact : Mrs. Jan Joseph – Executive Secretary L’art de Nok couvre environ 500 kilomètres, au nord du confluent du Niger et de la Bénoué sur le plateau de Jos. C’est à la civilisation de Nok que l’on doit les plus anciennes sculptures en terre cuite connues en Afrique subsaharienne. Ces merveilles de l’art antique du Nigéria ont été datées entre 600 avant J.-C et 300 après J.-C. Les statues ont une beauté classique et représentent des rois, des devins et des prêtres. Leur taille varie depuis les amulettes miniatures de 10 cm de hauteur jusqu’aux sculptures monumentales assises ou agenouillées dans des poses royales Nok art is found in an area some 500 km to North of the rivers Niger and the Benue, on the plateau of Jos. Made of baked clay, thoses sculptures wich are the oldest known in Subsaharian Africa characterize the Nok civilisation. These wonders of Antique art from Nigeria,were dated between 600 before B.C. and 300 A.D. The statues have a classical beauty and represent kings, soothsayers and priests. Their size vary between the 10cm miniature amulets and the monumental sitting or kneeling sculptures in royal poses.
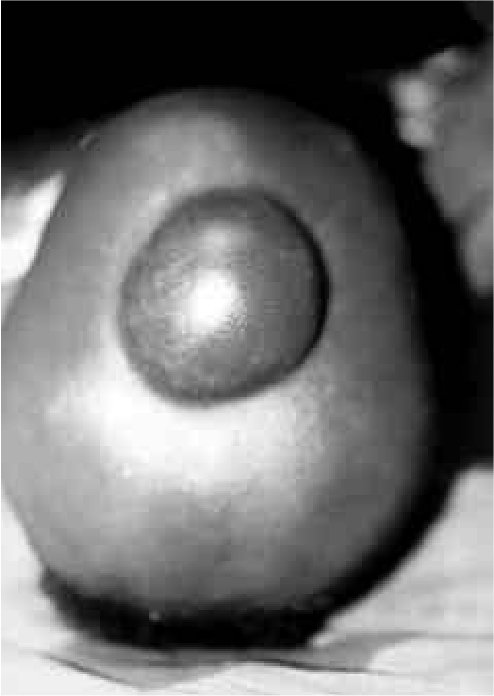
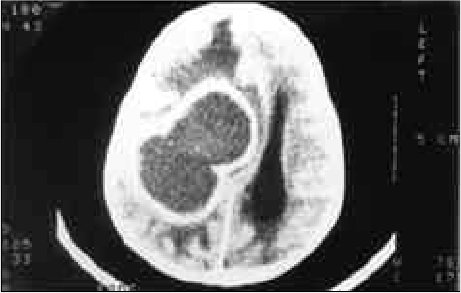
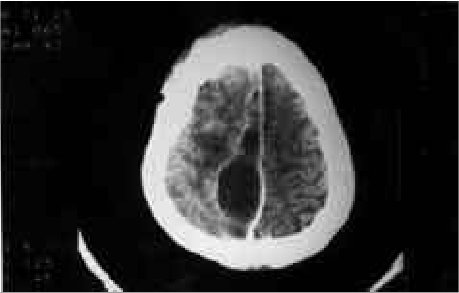
RESUME Description Objectif Méthodes Résultats Conclusion Mots clés : Afrique, Fontanelle antérieure, Kyste dermoïde crânie ABSTRACT Background Objective Methods Results Conclusion Keywords: Africa, Cranial dermoid cyst, Anterior fontanel INTRODUCTION Le kyste dermoïde, tumeur congénitale en règle unique et bénigne représente 0,1 à 2 % de toutes les tumeurs crâniennes (6,22). Dans la littérature, le kyste dermoïde est fréquemment décrit au niveau de la fontanelle antérieure (1 ,6 ,9 ,11 ,13 ,19 ,21 ,22). Les autres localisations sont plus rares (6,11). OBSERVATION Notre série comporte 5 patients (4 filles, 1 garçon) (Tableau et Figure) DISCUSSION Le kyste dermoïde résulte de l’inclusion aberrante d’éléments ectodermiques lors de la fermeture du tube neural entre la 3ème et la 5ème semaine du développement embryonnaire et représente 10% de toutes les masses extra-crâniennes (3). Il est de plus en plus admis que cette lésion expansive se rencontre dans toutes les races (7, 12, 13, 21, 22) avec cependant une très probable prédominance chez les africains (13). La notion de fréquence selon le sexe n’est mentionnée que par peu d’auteurs dans la littérature. Selon Adeloye (1), cette lésion surviendrait le plus souvent chez la femme avec un ratio 2/1. Peter ne fais pas la même constatation sur une série de 35 enfants, il a 20 garçons et 5 filles. Dans notre série, nous avons relevé comme Adeloye (1) une forte proportion de cas de sexe féminin, 4/1. L’échographie montre à la fois le contenu kystique et son caractère extra-dural, posé sur le sinus logitudinal supérieur dont l’aspect de triangle inversé à base supérieure est facilement reconnaissable. Dans la majeure partie des cas cet examen suffit (17). L’extension intracrânienne de ces lésions est visualisée à l’IRM. Nos constatations d’un point de vue histopathologique, confirment celles de tous les auteurs par l’existence de lésions kystiques très hétérogènes pouvant contenir de la kératine, du cholestérol, de la graisse, des poils, des sécrétions sébacées et sudoripares. Le liquide tumoral peut être clair, citrin ou xanthochromique en rapport avec un éventuel traumatisme sur la tumeur. Devant le tableau clinique d’un enfant présentant une tuméfaction de la voûte crânienne, médiane, un certain nombre d’affections doivent être discutées. Si les céphalhématomes, les lipomes, le sinus péricrânii, les malformations angiomateuses et le kyste osseux peuvent, de façon relativement aisée, être distingués du kyste dermoïde, il faut par contre recourir aux examens complémentaires, la tomodensiométrie cérébrale ou l’IRM notamment pour infirmer le diagnostic de céphalocèle ou de tumeur intracrânienne extériorisée. Le lipome dysraphique situé au niveau de la fontanelle antérieure bien qu’assez exceptionnel et s’accompagnant d’un lipome du corps calleux fait parti du diagnostic différentiel (4). Au plan anatomopathologique seule l’histologie peut différencier le kyste dermoïde du kyste sébacé (23). L’évolution spontanée du kyste dermoïde se fait vers un accroissement volumétriqueprogressif. Le traitement du kyste dermoïde est donc chirurgical. L’exérèse complète évite la survenue de récidive. La ponction du kyste avant la chirurgie est déconseillée car il y a un danger d’infection et le kyste se reconstituera inéluctablement (1). Au plan technique, on pratique une incision linéaire centrée sur la tumeur en évitant de l’effondrer avant son extirpation. Le décollement du tissu sous-cutané de la capsule tumorale est aisé. S’il existe une attache durale, on procède à une ligature suivie d’une coagulation. La seule difficulté signalée par certains auteurs est l’adhérence, rare, du kyste à la dure mère recouvrant le sinus longitudinal supérieur et qui empêcherait une exérèse complète compte tenu du risque hémorragique (17).
TABLEAU 1 : NOS OBSERVATIONS
RESUME Introduction Objectif Méthode Résultats Conclusion Mots clés : Abcès cérébral, Afrique, Empyème intracrânien, Enfant. ABSTRACT Background Objective Methods Results Conclusion Keywords: Africa, Child, Cerebral abscess, Intracranial empyema INTRODUCTION Les abcès et les empyèmes intracrâniens sont des pathologies fréquentes de l’enfant [1, 2]. Cette prédominance infantile semble plus nette dans les pays en voie de développement où les conditions socio-économiques défavorables constituent un facteur favorisant de ces pathologies. Dans ces pays, en raison du manque de moyens diagnostiques (tomodensitométrie et service de Neurochirurgie) ces pathologies sont très peu étudiées [1, 2, 11, 15, 16, 18]. En Côte d’Ivoire hormis l’étude de N’GOAN [19] ayant porté sur l’échographie transfontanellaire dans les méningites purulentes de l’enfant à Abidjan aucune étude ayant trait aux collections suppurées intracrâniennes n’a été réalisée chez l’enfant à la différence des adultes pour lesquels nous disposons de données récentes [3]. MATERIELS ET METHODES Nous avons effectué une étude rétrospective effectuée dans le service de neurochirurgie du CHU de Yopougon sur une période de 5 ans (de 1994 à 1998). Elle a porté sur 34 observations cliniques d’enfants dont l’âge variait entre 7 mois et 15 ans. Les aspects épidémiologiques, cliniques, thérapeutiques et le devenir des enfants après un recul de 2 à 6 ans ont été analysés. RESULTATS 1 – Données épidémiologiques Nous avons observé 12 cas (35,3 %) d’abcès contre 16 cas (47,05 %) d’empyèmes. L’association abcès et empyème a été constatée dans 6 cas (17,6 %). Une ostéite de la voûte crânienne a été observée dans 5 cas. Parmi les 16 cas d’empyème aucun cas d’empyème extra dural n’a été observé. L’âge moyen des patients était de 11 ans avec des extrêmes de 7 mois et 15 ans. Il a été constaté chez le grand enfant 22 cas de suppurations intracrâniennes, 10 chez le petit enfant et 4 chez le nourrisson. La prédominance masculine était nette avec un sex-ratio de 2/1. La porte d’entrée retrouvée dans 24 cas était ORL dans 13 cas dont 4 cas d’otite et 9 cas de sinusite, post méningite dans 6 cas, infection dentaire (3 cas) et cutanée (2 cas). La sérologie HIV réalisée dans 12 cas est revenue négative. Une hyperglycémie a été retrouvée chez deux patients. L’étude bactériologique a permis d’isoler des coccigram positifs (3 cas), entérobactéries (2 cas) streptocoque (2 cas) une flore mixte aéro-anaérobie (1 cas) et une association pseudomonas acinétobacter (1 cas). Dans les 23 autres cas ( 67,6 %) le germe n’a pas été isolé. 2 – Données cliniques et tomodensitométriques Les signes d’appel les plus couramment rencontrés étaient la fièvre (77,8 %) suivie des céphalées (75 %). L’hémiparésie (33,3 %) venait en 4ème position après les crises convulsives (36 %). L’altération du niveau de la conscience a constitué le signe d’appel dans 10,4 % des cas et les signes méningés 8,3 %. A l’examen physique, le syndrome prédominant était le syndrome pyramidal hémicorporel (64 %) suivi des troubles de la conscience avec un score de Glasgow moyen de 8 et des extrêmes allant de 4 à 12 (47,2 %), et du syndrome méningé (41,6 %). La triade de Bergman était présente 32 fois (94 %). Dans 13 cas (38,2 %) une suppuration du scalp a été retrouvée et dans 8 cas (26,4 %) un abcès palpébral. Le fond d’il n’a pas été fait systématiquement. 3 – Données thérapeutiques et le devenir des enfants traités d’abcès ou d’empyèmes intracrâniens La durée d’hospitalisation qui allait de 1 à 90 jours a permis d’enregistrer deux (2) cas de décès dont l’un à l’arrivée et l’autre un jour après. Ces deux (2) cas de décès étaient imputables à un engagement cérébral consécutif à l’hypertension intracrânienne. L’évolution a été favorable chez 32 patients avec des séquelles retrouvées dans 8 cas à type d’hypoacousie (1 cas), de retard du développement psychomoteur (1 cas) et d’épilepsie partielle secondairement généralisée (1 cas). L’épilepsie secondairement généralisée survenue deux mois après l’intervention chirurgicale était due à l’arrêt du traitement antiepileptique. L’hydrocéphalie a été retrouvée 4 fois : tétraventriculaire (3 cas) et triventriculaire (1 cas). Aucun cas d’aphasie ou d’hémiparésie n’a été constaté. De même il n’y a pas eu de récidive de la suppuration; le traitement de la porte d’entrée ayant été systématique. La durée moyenne de l’antibiothérapie était de 45 jours et celle de l’anti épileptique de 18 mois. DISCUSSION Au plan nosologique, parmi les trois entités de suppurations intracrâniennes les empyèmes sous duraux ont prédominé dans cette étude. Ceci marque une différence avec d’autres études où les abcès cérébraux sont prédominants avec une proportion de 4 empyèmes subduraux pour 3 abcès. Ainsi ALLIEZ a retrouvé 44 cas d’abcès contre 16 cas d’empyèmes sous duraux et 4 cas d’empyèmes extra duraux [2]. PONSOT sans donner de chiffre affirmait que les abcès étaient les plus fréquents des collections suppurées intracâniennes [20]. GUEYE et ses collaborateurs ont diagnostiqué 41 cas d’abcès contre 21 cas d’empyèmes chez les patients de sexe masculin et 14 cas d’abcès contre 4 cas d’empyème chez des patients de sexe féminin [11]. NATHOO a diagnostiqué 699 cas d’empyème sous dural contre 82 cas d’empyème extra dural et 712 cas d’abcès intracrânien [18]. BISSAGNENE a diagnostiqué en 8 ans, 19 cas d’abcès et 7 cas d’empyème [2]. Cette étude retrospective de 34 observations de suppuration intracrânienne confirme quelques données actuelles de la littérature. En effet le maximum d’abcès ou d’empyème intracrânien est retrouvé chez le petit et le grand enfant. Chez le nouveau-né et le nourrisson ces collections suppurées intracrâniennes sont exceptionnelles [14, 17]. Ces résultats concordants des différents auteurs s’expliquent par le fait que la porte d’entrée souvent ORL prédisposent le grand enfant et le petit enfant. Les cas de suppurations intracrâniennes observées chez le nourrisson et le nouveau-né sont secondaires à une méningite purulente [7, 8], une cardiopathie cyanogène [14], la pose d’une erfusion sur le scalp [12] et à une septicémie [20]. Tous les auteurs s’accordent à reconnaître également que le sujet de sexe masculin est le plus touché avec un sex-ratio de 3/2 [14] et 2/1 dans notre étude. Le maximum de suppurations intracrâniennes constaté chez le petit enfant et le grand enfant s’explique par la porte d’entrée qui est souvent ORL[3, 5, 11]. L’étude bactériologique du prélèvement de pus est diversement appréciée selon les auteurs. Nos résultats sont concordants avec ceux de certains auteurs [2, 14]. Pour LEYS et PETIT[14], actuellement il semble que dans plus de 50 % des cas le pus est stérile et dans d’autres cas il est polymicrobien [15]. Pour KORINEK [13] dans plus de 90 % des cas la culture du pus permet d’isoler le ou les germes. L’hémophilus influenzae, le pseudomonas et les entérobactéries retrouvés dans cette étude sont rarement à l’origine de suppuration intracrânienne [14]. Dans notre étude il a été retrouvé dans un cas d’abcès, une association acinétobacter et pseudomonas aéruginosa. L’étiopathogénie des suppurations intracrâniennes fait intervenir divers mécanismes à savoir une infection de voisinage, un traumatisme crâniofacial, une métastase à partir d’un foyer infectieux, un mécanisme idiopathique [5, 7, 9, 10, 16, 18, 21]. Dans ce travail les abcès et empyèmes intracrâniens ont été secondaires à la diffusion d’une infection de voisinage (ORL et méningée) et métastatique d’origine dentaire et cutanée. Les autres cas dont le mécanisme n’a pas été retrouvé pourraient être considérés comme idiopathique. Il n’a pas été constaté d’abcès ou d’empyème post traumatique. De même les cardiopathies cyanogènes responsables de la majorité des abcès cérébraux n’ont pas été observées [14]. Le diagnostic de suppuration intracrânienne peut être évoqué devant une triade de Bergman retrouvée ici dans 94 % des cas. Cependant les signes cliniques sont rarement au complet. Aussi une épilepsie focale dans un contexte fébrile, une hypertension intracrânienne rapidement évolutive doivent-ils attirer l’attention . Cette éventualité a été constatée dans 4 cas. La tomodensitométrie crânio encéphalique sans et avec injection intraveineuse de produit de contraste permet le diagnostic dans la majorité des cas. Lorsqu’elle est normale, l’imagerie par résonance magnétique nucléaire est l’exploration de choix tant pour le diagnostic que pour le suivi thérapeutique. Elle permet un diagnostic très précoce [7] car elle permet une meilleure différenciation et une meilleure appréciation de l’infection dans l’os et les tissus mous. Les empyèmes et abcès intracrâniens gardent une réputation de gravité à l’origine d’une lourde mortalité. De ce fait ils constituent une urgence médico-chirurgicale. L’antibiothérapie est instituée sans attendre l’isolement du germe. Nous avons utilisé une triantibiothérapie associant céphalosporine de 3ème génération, une aminoside et le métronidazole, ou une biantibiothérapie associant une céphalosporine de 3ème génération et le chloramphénicol. La péfloxacine a été utilisée chez le grand enfant âgé de 15 ans. L’utilisation d’antiépileptique systématique a permis dans notre pratique de réduire les séquelles épileptiques. Quant aux anti-démateux tels que le mannitol, les corticoïdes et le glycérol, leur utilisation n’a pas été systématique. Ils ont été réservés aux cas d’dème menaçant. Le traitement chirurgical a été nettement simplifié avec de bon résultat. L’exérèse d’abcès n’est plus pratiquée [7]. Actuellement les abcès sont ponctionnés [7, 10] par un trocard de Cushing à partir d’un trou de trépan lorsque l’indication chirurgicale est posée. Les empyèmes sous-duraux ont été exclusivement évacués après avoir réalisé une rondelle osseuse. Notre avis rejoint celui de BOK (4) et DECHAMBENOIT (6) en proposant l’utilisation préférentielle de la tréphine ou le trou de trépan pour le traitement de ces collections suppurées intracrâniennes [4, 6] contrairement à la taille systématique d’un large volet autrefois recommandé [15]. CONCLUSION Les abcès et empyèmes intracrâniens constituent une pathologie fréquente de l’enfant. Ils sont souvent secondaires à une complication d’une infection ORL. L’avènement de la tomodensitométrie permettant un diagnostic précoce et une prise en charge urgente à contribué nettement à l’amélioration du pronostic de ces collections suppurées intracrâniennes. Actuellement la tendance chirurgicale est à la simplification du geste. Le coût élevé du traitement, de 1231 $ en moyenne, insupportable pour nos populations généralement pauvres justifie la nécessité d’une prophylaxie efficace. Cette prophylaxie consiste en un traitement correct des infections ORL, dentaires, des méningites et le parage des plaies crânio encéphaliques.
Mots clés : Afrique de l’Ouest, Neurochirurgie, Stéréotaxie Keywords: Neurosurgery, Stereotactic surgery, West Africa INTRODUCTION The introduction of CT scanners into West Africa in the last decade has upgraded the practice of neurosurgery in the subregion. The CT scanner provides a three dimensional database of the brain. The combination of the CT scanner and stereotaxy optimises the use of the former. Stereotactic instrumentation provides the capability for rapid access with great accuracy to virtually any intracranial point. The rationale for applying stereotactic methodology to neurosurgical procedures is to access targets accurately with a minimum of spatial error (i.e. low bias) and a high degree of reproducability (i.e. high precision ). In the management of intracranial lesions, intracranial access can be achieved and valid answers obtained regarding processes that required craniotomy or high risk cerebral transit. The histological nature of any suspicious intracranial lesion may be determined safely and accurately thereby enabling the compilation of an accurate database for intracranial lesions. It is also possible to evacuate intracranial hematomas; aspirate cysts or abscesses; provide guidance for small lesions; perform functional procedures (eg. for movement disorders, epilepsy) and the implantation of interstitial radioactive sources into intracranial neoplasms. MATERIALS AND METHODS 17 patients (11F, 6M) with CT disclosure of intracranial mass lesions that could be assessed or managed stereotactically to the patients benefit (3,4) underwent stereotactic procedures during a consecutive 18-month period. Analysis of charts, relevant imaging studies and pathology reports were done. – CT-Guided Stereotactic Procedure. Patients were then transported to the OR suite where under sterile conditions and utilizing local anesthesia and IV sedation, intracranial access was obtained via a 2mm or 4mm twist drill calvarial opening. 3-6 biopsy specimens were taken in each case and subjected to histopathologic analysis. The safest and usually shortest transit penetrating one pial surface was utilized while taking into consideration the structures in the transit path to the lesion. Biopsy specimens were obtained with a Blaklund spiral biopsy needle. Facilities for intraoperative frozen section were not available. RESULTS 17 consecutive stereotactic procedures were performed. There were 6 males and 11 females; ages ranged from 2 to 72 years with one patient under 10 years. The mean age was 36.7 years (SD, 19.4). 16 cases (94%) achieved a definitive diagnosis, and 1 case was classified as failed biopsy, providing a failed biopsy rate of 6%. The pathological findings of the overall series are presented in table 2. – Imaging Characteristics – Pathological Findings, Management Arachnoid cysts were biopsied and aspirated in 2 patients. One of the patients who was 2 years at the time of the procedure, had complete resolution of the cyst confirmed by CT imaging performed 11 months after the stereotactic procedure. She has however developed non-communicating hydrocephalus for which endoscopic third ventriculostomy and or aqueductoplasty is planned. In total, 6 of the cases (35%) underwent both biopsy and aspiration/evacuation. – Cost The following assumptions were made a) no stereotactic scan is required for conventional craniotomy b) for stereotactic procedures, no days were spent in the Intensive/Critical care unit c) 2-3 days of hospital stay is required for stereotactic procedures as compared to 7-10 days for conventional craniotomy. It was found using the above parameters and assumptions that stereotactic procedures cost 50-59% less than conventional craniotomy. In the series, the average OR time was 37 minutes (R 30-43; SD 4). The mean duration of hospital stay was 2.7 days (R 2-7days; SD 1.4). DISCUSSION Two old and simple concepts, a three-dimensional positioning stage and a coordinate system were combined in 1906 to create a new one; the stereotactic method (7). The advent of computer-based medical imaging applied to the stereotactic method encouraged the adaptation of the later to the management of intracranial tumors (13). The incorporation of CT scanning into stereotactic technique in the early 1980’s was an obvious step for two reasons. First, CTprovided a precise 3-D database that can be readily translated into the 3-D coordinate system of a stereotactic frame. Second with CT scanning, tumors could be seen directly instead of having their positions inferred from shifts of components of the ventricular system or from shifts of vessels on cerebral angiography. Evaluation of the enhanced anatomic detail provided by the CT scanner could therefore be used for surgical planning. The availability of stereotactic surgical equipment and technique therefore optimises the use of the CT scanner. Optimal use of a CT scanner is crucial since the acquisition and maintenance of a CTscanner involves a large capital investment. Our study reveals that the mean CTscan time of 18 minutes required for stereotactic data acquisition can readily be fitted into the schedule of a Radiology Department in the region without significant disruption. Appuzo et al reported that a scan utilization time of less than 15 minutes renders the issue of scanner access in a busy neurosurgical service inconsequential (1). Furthermore, the acquisition of MRI equipment by an institution in West Africa will be superfluous unless that institution has the capability to perform stereotactic surgery. Hitherto, localization methods for intracranial procedures in West Africa have been qualitative and imprecise. Large skin and bone flaps have been utilized in order to ensure that the relevant lesion lay within the limits of the craniotomy. Consequently, general anesthesia, blood transfusions and several days of critical post-operative care have been required. The lack of relative availability of these resources within the sub-region has made the practice of neurosurgery difficult and problematic. The purpose of incorporating stereotactic methodology into neurosurgical practice is to provide an improvement in localization over that which is available. The proper clinical use of stereotactic methodology, requires a mature technological understanding of the available instruments and a clear understanding of their benefits and limitations. Clinically, the determinants of application accuracy should be considered before every use of stereotactic methodology for any therapeutic intervention (11,18). As shown from this study, stereotactic procedures can be accomplished without the need for general anesthesia, blood transfusions and critical post-operative care. An added advantage is the ability to surgically treat neurologic patients with mild or severe systemic disease that is incapacitating or life-threatening (ASA II-IV) with much less added risk. The average OR time of 37 minutes is much less than that required for a conventional craniotomy. A comparative cost analysis revealed a 50-60% reduction in total costs for stereotactic procedures when compared to conventional craniotomy. This analysis took into account the cost of the stereotactic CT scan, operating room costs, pharmaceuticals and length of hospital stay. The mean duration of hospital stay of 2.7 days is considerably lower than for conventional craniotomy. In the sub-region almost all patients who undergo craniotomy have had to remain in hospital for at least 7 days in order to ensure proper wound healing and suture removal before discharge. Stereotactic procedures also reduce the need the OR swabs/sponges, patties, scalp clips, sutures, wound drains, wound dressings and laboratory tests. Further cost reductions are obtained by reducing the need for Intensive/Critical care and the utilization of local anesthesia complemented by intravenous sedation for procedures. Stereotactic procedures will help to reduce the on health care professionals and resources in the sub-region. All the target lesions were in the supra-tentorial compartment. However, stereotactic procedures can also be performed for posterior fossa lesions (1,8,12,20). Table 2 gives the histopathologic processes substantiated in the 17 biopsied lesions. The diagnostic biopsy rate has varied between 91 and 100% (2,9). Diagnostic success is predicated on proper case selection, precise point target tissue retrieval and informed pathologist feedback. Proper case selection demands that the decision to employ stereotactic biopsy should be preceeded at all times by a thorough neurologic and radiographic assessment of the patient. Lesions such as ischemic infarcts, vascular malformations and multiple sclerosis should not be biopsied. The prevention of targeting error can be technically achieved by careful data entry and the avoidance of angulation of biopsy probes at the calvarial entry point. Real-time intraoperative imaging can in the future provide for monitoring the biopsy needle in relation to the intended target. Finally, the availability of intraoperative frozen section/smears often provides useful information to guide the surgeon and should be used whenever possible (21). We had to depend entirely on review of histologic sections after permanent fixation; facilities for frozen section review are not available. There is a wide range of failed biopsy rates in the literature, 3-47%. This is as a result of the wide differences in definition of failed biopsy rates (1, 5, 10, 15-17, 19, 22-24). Soo et al have classified failed biopsies as lesional or nonlesional (24). Lesional failed biopsies reflect a nonspecific pathologic change e.g astrogliosis, necrosis or inflammatory change. Lesional failed biopsies can be further divided into representative and nonrepresentative. The representative group have a time window outside which definitive diagnosis cannot be made as the pathologic elements become less distinct e.g radionecrosis, subacute infarction or a demylinating plaque from multiple sclerosis. In these situations it is sometimes impossible to obtain a definitive diagnosis even after craniotomy and open biopsy. Although these lesions are generally not biopsied, in some instances, neither clinical judgement or current available imaging modalities can differentiate them from neoplastic or infective processes, hence necessitating biopsy. The lesional nonrepresentative failed biopsies are due to biopsying either the reactive edges or the necrotic areas of hetrogenous neoplastic or infectious processes. These may be considered as a relative target selection error or minor targeting error. Inspite of the limited diagnostic usefulness of lesional failed biopsy, in certain cases the pattern of the changes suggests a specific diagnosis such as tumor necrosis versus coagulative necrosis of infarction. In nonlesional failed biopsy, the predetermined target on a static CT scan was missed, yielding normal brain. This may occur as a result of the lesion migrating away from an advancing biopsy needle. A slight angular deflection of the semirigid biopsy needle on account of an angled twist drill calvarial entry may lead to the biopsy device missing the lesion tangentially. The failed biopsy rate for the series is 6%. Metaanalysis of 9,467 published cases of stereotactic biopsy from series with over 100 cases yields a failed biopsy rate of 9% (24). It has been commonplace to blame diagnostic failure on the size of the biopsy specimen, i.e it is too small (14). However a representative specimen is always of sufficient size for a diagnosis to be made and representative tissue is better obtained with stereotactic technique than with open biopsy methods (6). The elucidation of the molecular pathogenesis of CNS tumors will hopefully lead to a to a molecular classification and enable improved diagnostic yields from small stereotactic biopsy specimens, eg DNA analysis using polymerase chain reaction requires less than 100ng of DNAto identify infectious agents such as toxoplasma; differntiation of astrogliosis from a low grade glioma using molecular markers such as p53 mutation, loss of genetic information on chromosome 19q or over expression of growth factor receptors implicated in tumorigenessis (24). From an initial start of biopsies and aspirations/evacuations, a stereotactic surgery program in West Africa can be expanded to include stereotactic endoscopy, stereotactic craniotomy, functional neurosurgical procedures and radiosurgery. The benefits from such a program will include, the acquisition of an accurate histologic database for intracranial lesions, capability for neurophysiologic research, enhanced medical education for medical students and neurosurgical residents, clinical improvements in patient care and reduction of health care costs. Table1 Location of target masses (17 procedures)
Table2 Summary of histologic diagnosis in 17 stereotactic biopsies
Articles récents
Commentaires récents
Archives
CatégoriesMéta |




© 2002-2018 African Journal of Neurological Sciences.
All rights reserved. Terms of use.
Tous droits réservés. Termes d'Utilisation.
ISSN: 1992-2647