
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
INTRODUCTION L’amyotrophie spinale proximale ou spinal muscular atrophy (SMA) est une maladie neuromusculaire autosomique récessive causée par des délétions ou des mutations du gène SMN1 situé sur le chromosome 5q11.2 à 13.3 (17). Elle est la principale cause génétique de mortalité infantile, affectant une naissance vivante sur 10 000 (29). La gravité de l’amyotrophie spinale est déterminée en partie par le gène SMN2 localisé à côté du gène SMN1 sur le chromosome 5q. Le nombre de copies de SMN2 détermine aussi le type d’amyotrophie spinale, les patients atteints de SMA de type I ayant le plus souvent 2 copies de SMN2, les patients de type II ont 3 copies et les patients de type III peuvent avoir 4 copies ou plus (5). La meilleure compréhension de la physiopathologie et le développement de la thérapie génique ont permis la découverte de nouvelles molécules contre cette maladie. Ainsi ces dernières années, 3 médicaments ont été approuvés par la US Food and Drug administration (FDA) dans le traitement de l’amyotrophie spinale : le Nusinersen (Spinraza®) en décembre 2016, le onasemnogene abeparvovec (Zolgensma®) en mai 2019 et le risdiplam (Evrysdi®) en aout 2020. L’European Medecines Agengy (EMA) les autorise elle aussi quelques mois plus tard (7). Nous ferons ici le point sur ces trois molécules et citerons les thérapeutiques en cours d’essai. PHYSIOPATHOLOGIE La SMA est causée par une anomalie génétique dans le gène du motoneurone de survie SMN situé sur le chromosome 5 (17). Chez l’homme, ce gène existe sous deux formes presque identiques, le gène SMN1 télomérique et le gène SMN2 centromérique. La principale différence fonctionnelle entre ces deux gènes est liée à un seul changement de nucléotide au début de l’exon 7, la cytosine étant remplacée par une thymidine dans le gène SMN1. Cette transition de C à T modifie un modulateur d’épissage exonique, ce qui entraîne le saut fréquent de l’exon 7 pendant la transcription de SMN2 (3,10). Le gène SMN2 tronqué au niveau de l’exon 7, ne permet pas de compenser l’absence de protéine SMN dans les cellules. Néanmoins, le nombre de copies du gène SMN2 variant selon les patients détermine le phénotype clinique de la maladie par l’expression d’une quantité variable de faibles niveaux de protéine SMN fonctionnelle. L’absence de la protéine va entrainer la dégénérescence du motoneurone alpha de la moelle épinière et du tronc cérébral, entraînant une faiblesse et une atrophie des muscles squelettiques des membres et du tronc, ainsi que des muscles bulbaires et respiratoires (27). Il existe une relation inverse entre le nombre de copies de SMN2 et la sévérité́ de la maladie. (2,15). TRAITEMENT La compréhension du mécanisme génétique sous-jacent de la SMA a conduit au développement de deux thérapies ciblées qui augmentent la production de la protéine SMN fonctionnelle, la première en modifiant l’ARN SMN2 pour produire une protéine SMN complète (nusinersen et risdiplam) et la seconde par l’administration directe du gène SMN1 via un vecteur viral (onasemnogene abeparvovec) (13). Leurs mécanismes d’action, voies d’administration et leurs coûts sont résumés dans le tableau I (7). Nusinersen Premier médicament autorisé pour la SMA, le Nusinersen est un oligonucléotide antisens qui se lie à un site intronique d’épissage-silence dans l’intron 7 du SMN2 et inhibe l’action d’autres facteurs d’épissage, favorisant l’incorporation de l’exon 7 dans l’ARNm. Ce mécanisme permet la traduction d’un niveau plus élevé de protéine SMN fonctionnelle avec une amélioration significative de la survie (16,22). Sur la base de ces données, des essais cliniques ont été lancés et les sujets ont toléré en toute sécurité de multiples injections intrathécales en phase II, avec des preuves d’amélioration de la fonction motrice. Les données de phase III réussies ont conduit à l’approbation de la FDA et de l’EMA en décembre 2016 et juin 2017, respectivement, comme premier médicament pour traiter les patients atteints de SMA de type 1-3 5q (11). Les nourrissons traités avant 6 mois ou plus tard, dans les essais ENDEAR et CHERISH, respectivement, ont montré des résultats positifs en termes d’étapes motrices et de survie sans événement ; par conséquent, les deux essais de phase III ont été interrompus au stade intermédiaire pour permettre à tous les participants de passer au nusinersen dans une étude ouverte (SHINE) (12). Le nusinersen est indiqué dans tous les types de SMA, quel que soit leur âge et est administré par voie intrathécale avec 4 doses de charge de 12 mg en 2 mois et des doses d’entretien de 12 mg aussi tous les 4 mois (5,7,23). Des complications peuvent résulter de la ponction lombaire, notamment de rares cas d’hydrocéphalie et des céphalées. La ponction lombaire peut également être difficile chez les patients souffrant de scoliose ou de fusion vertébrale, ce qui nécessite l’utilisation d’un guidage basé sur l’imagerie interventionnelle et des cathéters intrathécaux (13,16). Les effets secondaires qu’on peut rencontrer au cours du traitement sont la thrombocytopénie, les anomalies de la coagulation et la toxicité rénale. La numération plaquettaire, les études de coagulation et les analyses d’urine sont contrôlées au départ et avant chaque perfusion (23). Dans la SMA infantile, il a été démontré que le nusinersen réduit de moitié le risque de décès ou de ventilation mécanique permanente (11). Le bénéfice était le plus important chez les enfants traités de manière pré-symptomatique ou dans les 3 mois suivant l’apparition de la maladie. Le nusinersen s’est également avéré bénéfique chez les enfants plus âgés dont la SMA était apparue après l’âge de 6 mois et qui ont été traités entre 2 et 12-15 ans (12,20). Onasemnogene abeparvovec Médicament le plus cher au monde (2 millions de dollars la cure), l’onasemnogène abeparvovec est une thérapie génique conçue pour délivrer une copie entièrement fonctionnelle du SMN humain afin de traiter la cause génétique de l’amyotrophie spinale infantile en une seule perfusion. Il utilise un vecteur de virus adéno-associé de sérotype 9 (AAV9) pour exprimer une construction SMN recombinante auto-complémentaire humaine sous le contrôle d’un activateur hybride du cytomégalovirus et d’un promoteur de la β-actine de poulet (8). Il est indiqué dans les SMA de type 1. Les patients traités dans le cadre de l’essai clinique qui a conduit à son approbation étaient tous atteints de SMA de type 1, avec 2 copies de SMN2, et âgés de moins de 8 mois (19). Ce premier essai a traité 15 nourrissons, 3 à faible dose et 12 à forte dose : les 15 patients ont tous survécu jusqu’à 20 mois sans avoir besoin d’une assistance respiratoire, 11 d’entre eux ont acquis la position assise sans assistance et 2 marchant même de manière autonome (19). La FDA l’a autorisé pour le traitement des enfants jusqu’à l’âge de 2 ans. L’approbation européenne diffère légèrement, autorisant le traitement de tous les enfants de moins de 21 kg, quel que soit leur âge (14,19,23). Le traitement se fait en une perfusion unique de 1,1 1014 génomes vecteurs par kilogramme administrée en une heure. (5). Son Principal avantage est qu’il ne nécessite qu’une seule injection mais son cout élevé et sa synthèse complexe constituent un frein à son accès (4,22). L’étude STR1VE a inclus 22 patients atteints d’amyotrophie spinale de type 1 qui ont reçu l’onasemnogène abeparvovec. Treize (59 %, 97-5 % IC 36-100) des 22 patients ont acquis une assise fonctionnelle indépendante pendant 30 secondes ou plus lors de la visite d’étude à l’âge de 18 mois (vs 0 des 23 patients de la cohorte non traitée ; p<0-0001). Vingt patients (91%, 79-100) ont survécu sans ventilation permanente à l’âge de 14 mois (contre 6 [26%], 8-44 ; p<0-0001 dans la cohorte non traitée (8). Tous les patients ayant reçu l’onasemnogène abeparvovec ont présenté au moins un événement indésirable, le plus fréquent étant la fièvre (8,19). L’effet secondaire la plus redoutable est l’atteinte hépatique après l’administration du médicament, probablement en raison d’une réponse immunitaire au vecteur viral. L’adjonction quotidienne de stéroïdes (1mg/kg) pendant un mois et la surveillance des tests de la fonction hépatique ont permis d’éviter toute autre toxicité hépatique (13,19,21). Il a été aussi décrit des cas microangiopathie thrombotique parmi les effets secondaires à craindre (6). Risdiplam Le Risdiplam est une petite molécule qui module l’épissage du gène SMN2, en se liant à deux sites dans le pré-ARNm SMN2 : le site d’épissage 5′ (5′ ss) de l’intron 7 et l’exhausteur d’épissage exonique 2 (ESE2) dans l’exon 7. La spécificité unique de la liaison de deux sites augmente les niveaux de l’ARNm et de la protéine SMN de taille normale, tout en réduisant l’impact sur l’épissage d’autres pré-ARNm et en évitant la possibilité d’effets hors cible (22,25). Le risdiplam est autorisé par la FDA pour les types I, II et III en aout 2020 (28). La posologie recommandée de risdiplam est déterminée par l’âge et le poids corporel, avec une posologie de 0,2 mg/kg/jour recommandée pour les patients âgés de 2 mois à < 2 ans, de 0,25 mg/kg/jour recommandée pour les patients ≥ 2 ans pesant < 20 kg, et de 5 mg/jour recommandée pour les patients ≥ 2 ans et pesant ≥ 20 kg. Risdiplam doit être pris par voie orale une fois par jour après un repas, approximativement à la même heure chaque jour (9). Les résultats récemment publiés de l’étude de phase II/III FIREFISH ont démontré une meilleure efficacité à la dose testée la plus élevée, pour laquelle 7 des 17 nourrissons ont pu s’asseoir de manière indépendante après 12 mois de traitement. Une étude de suivi est en cours, portant exclusivement sur les effets à long terme de la dose plus élevée44. D’autres essais cliniques de phase III sont en cours, SUNFISH évaluant l’efficacité du risdiplam chez les patients de type II/III âgés de 2 à 25 ans, JEWELFISH étudiant les effets du risdiplam chez les patients précédemment traités par d’autres thérapies de la SMA et RAINBOWFISH étudiant le risdiplam chez les nourrissons pré-symptomatiques atteints de SMA âgés de moins de 6 semaines à la première dose (1,7). Les effets secondaires les plus courants dans les essais cliniques du risdiplam étaient la fièvre, les éruptions cutanées, les ulcères de la région buccale, les douleurs articulaires, la diarrhée et les infections des voies urinaires. La population de nourrissons recevant du risdiplam a présenté des effets secondaires supplémentaires, notamment une infection des voies respiratoires supérieures, une pneumonie, des vomissements et une constipation (28). Autres traitements En dehors des trois molécules déjà autorisées dans le traitement de la SMA, il existe d’autres molécules en cours d’essai clinque. Le reldesemtiv est un modulateur de l’activité de la troponine rapide, qui a montré un bénéfice dans la fonction motrice des patients atteints de troubles du motoneurone, dont la SMA, dans les premières études (26). Le SRK-015 est un anticorps monoclonal dirigé contre la myostatine. Dans un modèle murin de SMA, on a constaté qu’il augmentait la masse et la force musculaires après un traitement visant à restaurer l’expression de la protéine SMN (18). Le branaplam a été identifié à l’aide d’un crible à haut débit pour l’inclusion de l’exon 7 de SMN2 et semble stabiliser le pré-ARNm SMN2 avec des complexes de facteurs d’épissage.46 L’administration quotidienne a montré une augmentation dose-dépendante de l’inclusion de l’exon 7 et de l’expression de la protéine SMN chez les souris SMA, avec une amélioration du poids corporel et de la durée de vie (24). La liste est non exhaustive. COMMENTAIRES À l’heure actuelle, il n’existe pas de preuves suffisantes indiquant la supériorité d’un traitement sur les autres. L’âge d’apparition, l’état fonctionnel actuel, la scoliose/les fractures, le statut des anticorps AAV9 et le nombre de copies de SMN2 sont les principaux facteurs qui guident le choix du traitement Les données sur la thérapie combinée dans la SMA sont limitées et, jusqu’à présent, aucun essai clinique ne l’a étudiée directement pour les médicaments actuellement approuvés. Il existe peu d’études cliniques contrôlées chez les patients adultes atteints de SMA, ce qui limite la capacité à quantifier les gains attendus du traitement (14). Sur le plan éthique, la question d’accès aux soins pour tous est toujours soulevée par certains cliniciens vu les coûts assez élevés d’une part, les conséquences à long terme de la thérapie génique qui n’a pas encore livré tous ses secrets d’autre part. Avec la découverte de ces nouvelles thérapies, le dépistage des femmes enceintes et des nouveaux nés commence à être systématique dans certains pays. Cependant ce dépistage n’identifie que les délétions homozygotes du SMN1 et détecte donc environ 95 % des nourrissons atteints de SMA, par conséquent, elle passe à côté de 5 % des nourrissons atteints de SMA qui présentent une délétion hétérozygote et une variante de séquence pathogène (3). Vu les coûts assez élevés de ces traitements, les pays en voie de développement notamment les pays de l’Afrique subsaharienne n’ont pas encore accès à ces thérapeutiques. Cependant beaucoup de ces pays ne font pas encore la génétique de l’amyotrophie spinale pour confirmer le diagnostic et connaitre le nombre de copies de SMN2, préalable au choix du médicament à instaurer. CONCLUSION Alors que les traitements de la SMA continuent de progresser, les prestataires de soins devront redéfinir son histoire naturelle, adopter une approche plus proactive de la rééducation et de la gestion multidisciplinaire, et utiliser les biomarqueurs pour créer des approches thérapeutiques personnalisées pour leurs patients. En fin de compte, les traitements combinés et la rééducation peuvent être nécessaires pour optimiser les résultats de certains patients atteints de SMA, mais des recherches supplémentaires sont nécessaires pour déterminer s’il y a un avantage supplémentaire et, le cas échéant, quels patients ont besoin de traitements multiples.
Tableau 1 : Résumé des 3 thérapies approuvées dans la prise en charge de la SMA[] [7]
FACTORS INFLUENCING SURGICAL OUTCOMES OF CAUDA EQUINA SYNDROME IN TOGO INTRODUCTION The cauda equina syndrome (CES), characterized in its complete form by paraplegia, sphincter disorders, and saddle anesthesia is relatively rare. Its annual incidence is 5 to 10 cases per 1 million. It’s responsible of important long-term morbidity, why requiring urgent therapeutic measures (3,10,15,22,30). The treatment is based almost exclusively on surgery, which results are variously assessed in the international literature (7,8,17,29). The predictor’s factors influencing outcomes are controversial (14). In Africa, the specificities as long delays of treatment, the lack of all therapeutic modalities and the patients follow up difficulties are greatly influencing the results (26,29). This motivated the current study to try to find factors influencing surgical outcomes of CES in Togo. MATERIAL AND METHODS This was a descriptive retrospective study from 2012 to 2021 including all patients operated for cauda equina syndrome in the neurosurgery department of the CHU Sylvanus Olympio in Lomé. CES was defined as the partial or total association of the following 3 disorders: – Motor disorders: total or partial deficit affecting at least one of the roots below L1; – Perineal sensation impairment: hypoesthesia or anesthesia; – Genito-sphincter disorders: urinary and faecal incontinence, urinary emergency, incomplete urination, urinary retention, erectile disorder for men. The complete syndrome was defined as association of paraplegia, saddle anesthesia, and anal and/or urinary incontinence. In addition to those disorders, CES must be confirmed by the compression of nerve roots of cauda equina with imaging. We assessed surgical outcomes over 2 periods of time : short term (6 months postoperative) and middle term (12 months postoperative). The parameters were based on motor, sphincter and sensory recovery ; each of them can be total, partial or null. The results were : – Good : when the recovery of each of the 03 functions was total ; – Satisfactory : sphincter control associated with motor recovery more than 3 ; – Bad : in the other cases. Multi-variable analysis (R software) was used to determine predictors. The factors assessed concerning neurological recovery were age, sex, symptoms duration, etiologies and initial neurological status. Two-sided p values < 0.05 were considered statistically significant RESULTS A total of 56 patients were operated for CES during the study period. The average age was 49.7 years (18 to 79 years) with male predominance (sex ratio of 2.3). The onset was sudden in 26.8%, rapidly progressive (< 15 days) in 50% and progressive (> 15 days) in 23.2%. Patients presented clinical history of lumboradiculalgia in 51.8% with radicular claudication in 23.2%. All the patients had motor and sensory disorders and 92.8% genito-sphincter disorders (Table I). The symdrome was complete in 48.2%. The average time for progression of the syndrome before diagnosis was 12.7 days. Only 8.9% of patients consulted within 2 days of cauda equina syndrome onset. CT scan (48.2%) and MRI (82.1%) found mainly degenerative causes as the etiology of the syndrome (Table II). Two patients had been operated on within 24 hours and the majority within a week after admission (96.4%). We did after laminectomy, 19 discectomies, 17 osteosyntheses and 08 tumor resections including 3 total removals (neurinomas). Radiation therapy was performed for a case of chordoma, Chemotherapy for plasmacytoma and anti-tuberculosis. The mean length stay was 25.8 days. Functional rehabilitation was daily during hospitalization and after discharge, only 24 patients (42.9%) honored it. In the medium term, surgical outcomes on neurological recovery were satisfactory, especially for incomplete forms. The difference between the 2 forms was statistically significant for good and satisfactory results (p= 0.000). The recovery was also better for traumatic CES comparative to other etiology (p=0.04). The recovery was independent to the duration of evolution (Figure 1) as well as for sex (p=0.8) and age (p=0.8). (Table III). DISCUSSION CES is characterized by several sign making its definition difficult (1,2,7). Whatever the form, it is a surgical emergency. Early surgery upon diagnosis is an important parameter for neurological recovery; the ideal time, although controversial, would be surgery within 24-48 hours after onset, if not as soon as possible (5,13,20,32). As our patients were operated late, there was no difference in neurological recovery between according operating times. On the other hand, we noted a significant difference between the complete or not CES in the evolution in favor of the incomplete forms as also underlined by several authors (7,11,31,33). The other parameters influencing neurological recovery are the duration of the syndrome, etiology, age, sex and functional rehabilitation (16,19). Regarding the etiology, the post-traumatic causes would have a better neurological recovery than the others (12,31). We noticed it in our series. Our operative findings may could explain this fact: we noted in all cases of post-traumatic cauda equina syndrome, a dural rupture. The existence of this rupture would reduce the intrathecal pressure, but would also facilitate the mobility and spreading of the rootlets in the available intracanal space, resulting less stress on them. This is reflected in a mostly incomplete syndrome, resulting in better post-surgical recovery. This mechanism would not occur in cauda equina syndrome due to herniated disc in which the dura being intact, generates by compression, a significant intrathecal hydrodynamic pressure damaging on the rootlets (9,20,21,24,25,28). Age does not seem to influence neurological recovery as found in our series (19). But we believe that it could be if we choose the right comparison groups. Indeed, we believe that the presence of degenerative lesions with chronic vascular alterations could slow down recovery compared to the subjects free of these lesions. Although it was demonstrated no recovery difference between the 2 sexes ; in the long term, some authors have found that women have more urinary incontinence than men, solely due to the anatomical specificity of the sphincters of the 2 sexes (19). Concerning disease duration, it has been shown that the slow evolution augurs better neurological recovery, especially because it mostly giving an incomplete CES (6,11,19,27). The complete CES which often reflects a total interruption of the nerve impulse (anatomical or functional) is most often correlated with a sudden onset. It is in this case a rapid decompression is very important before occurrence of irreversible ischemia, because the Wallerian degeneration that follows compromises neurological recovery after surgery. The role of physiotherapy is well established (18). Our results could be better if there were intensive rehabilitation centers ; because only 43% of outpatients were able to perform 3 weekly physiotherapies during the follow-up period. However, our results may probably evolve favorably in the long term, as neurological improvements occurred later than 4 years (4). CONCLUSION This study reflected like many others the difficulty to identify the prognostic factors of CES even some of them are reaching a consensus such as the initial neurological status and the etiology. The speed of management, even though controversial, is also beneficial for the patient first to avoid to complete his syndrome but also to avoid irreversible ischemia of the root vessels which definitively compromises neurological recovery. We noted also that, despite long delays, our surgical outcomes are overall good.
Tables Table I : Signs of cauda equina syndrome at admission.
⃰ for male (39)
Table II : cauda equina syndrome etiologies
Table III : surgical outcomes according to different factors
1 The recovery is better (good and satisfactory results) for incomplete CES (p=0.000) 2 The recovery is independent concerning age (p=0.8) 3 The recovery is independent concerning gender (p=0.8) 4 The recovery is better (good and satisfactory results) for traumatic CES (p=0.04)
Figure 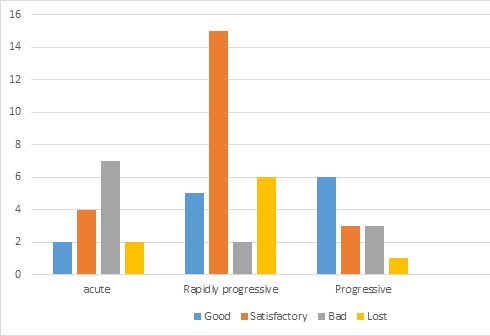 Figure 1 : Patients outcomes according to onset mode INTRODUCTION La chorée fait partie des syndromes hyperkinétiques des mouvements anormaux avec une prévalence de 5% de tous les mouvements anormaux (3). Les étiologies sont nombreuses et sont dominées par les causes infectieuses dans les pays sous-développés. La chorée de Sydenham est la plus fréquente des chorées aigues infectieuses de l’enfant avec parfois des séquelles motrices importantes (2). L’objectif de notre étude était de caractériser les aspects sociodémographiques, cliniques, paracliniques, thérapeutiques et évolutifs des enfants diagnostiqués de chorée de Sydenham au niveau de l’unité de neuropédiatrie du centre hospitalier national d’enfant Albert Royer. PATIENTS ET MÉTHODES Il s’agissait d’une étude rétrospective et descriptive allant de janvier 2005 à janvier 2019 qui s’est déroulée à l’unité de neurologie pédiatrique du Centre National Hospitalier d’Enfants Albert Royer (CNHEAR). Ont été inclus dans notre étude tous les patients suivis régulièrement pour chorée de Sydenham et dont l’âge était inférieur ou égal à 16 ans. Les patients dont les dossiers étaient incomplets ou inexploitables ont été exclus. L’étude des dossiers des patients suivis nous a renseigné sur les caractéristiques sociodémographiques, les données cliniques et paracliniques, les aspects thérapeutiques et évolutifs. L’analyse statistique des données a été réalisée grâce au logiciel Excel 2016. RÉSULTATS En 14 ans, nous avons colligé 11 patients. Le sexe ratio était de 0,57 avec une prédominance féminine (63,63%). L’âge moyen était de 9,45 ans et des extrêmes allant de 8 à 15 ans. La tranche d’âge de 6 à 10 ans était la plus représentée avec un pourcentage de 63,63%. (Cf. Tableau I). Sur le plan clinique, la chorée était généralisée chez 9 patients (81,81%) et localisée chez 2 patients (18,18%). Les formes localisées concernaient l’hémicorps droit (hémichorée). Seulement trois patients présentaient d’autres signes neurologiques à type de dysarthrie. Les signes extra-neurologiques étaient dominés par les cardiopathies (insuffisance mitrale isolée chez 6 patients et insuffisance mitrale plus aortique chez deux autres patients). Notons l’existence d’arthralgie chez 3 de nos patients. Sur le plan paraclinique le bilan infectieux était systématique avec un syndrome inflammatoire biologique non spécifique retrouvé chez 3 patients (élévation CRP et VS) et les ASLO positif (supérieur à 400) chez 7 patients. L’imagerie cérébrale n’était pas réalisée que chez deux enfants, la TDM cérébrale était normale chez les 7 enfants et l’IRM cérébrale n’avait objectivé que des hypersignaux bithalamiques et corticaux chez un patient sur les deux ayant bénéficiés de l’IRM. Tous les patients ont bénéficié d’un traitement à base de neuroleptique (halopéridol), trois d’entre eux ont reçu en plus de l’halopéridol (0,2 mg/kg/j), du clonazépam (0,05 mg/kg/j). Huit (8) des 11 patients ont bénéficié d’une corticothérapie à base de prednisone (2mg/kg/j) pendant 12 semaines. L’évolution était favorable chez 90,90% de nos patients diagnostiqués de chorée de Sydenham avec des délais de rémission allant de 1 à 12 mois et une moyenne de 4,27 mois. Un patient avait présenté une rechute après un arrêt précoce du traitement. L’ensemble de ces résultats est résumé dans le tableau II. DISCUSSION La chorée de Sydenham est la plus fréquente des chorées infectieuses de l’enfant (2). Sa prévalence importante dans les pays en voie de développement peut s’expliquer par les conditions d’hygiène défectueuses et le bas niveau socio-économique. L’amélioration du niveau de vie, l’avènement des antibiotiques, et les dispositions prophylactiques avaient rendu l’affection rarissime dans les pays développés (7). Cependant force est de reconnaitre la diminution de la chorée de Sydenham dans notre contexte. La nette prédominance féminine était retrouvée dans notre étude conformément aux données de la littérature (5,8). La chorée est le plus souvent localisée, soit à un hémicorps (hémichorée) soit à un segment de membre (monochorée) avec des fréquences de 10 à 20% (6,7). Dans notre étude la chorée était généralisée chez 81,81% des patients et les signes associés étaient dominés par les valvulopathies (4). Le traitement symptomatique de la chorée de Sydenham est de nos jours multiple. Certains médicaments comme les neuroleptiques halopéridol, pimozide et la chlorpromazine ont toujours montré leur efficacité avec une nette supériorité de l’halopéridol sur ces derniers. Cependant, l’existence des effets secondaires sont plus notés chez les patients sous halopéridol (1). Les antiépileptiques comme la carbamazépine et la valproate de sodium sont aussi utilisés dans le traitement symptomatique des chorées de Sydenham avec une supériorité des antiépileptiques par rapport aux neuroleptiques du fait de leur risque moindre d’effets secondaires. Dans notre étude les patients étaient tous sous halopéridol avec une bonne amélioration clinique. (1,9). Tous nos patients ont bénéficié d’une antibiothérapie à base de pénicilline. Une cure de 10 jours ou une injection en intramusculaire unique de pénicilline est recommandée même à l’absence de signe d’évolutivité d’infection streptococcique (10). Dans notre étude comme celle de la littérature, l’évolution est le plus souvent favorable après traitement et quelques rares cas de rechutes ou de persistances des chorées sont décrites et le plus souvent c’est l’inobservance thérapeutique ou l’existence d’une autre cause sous-jacente qui serait à l’origine (1). CONCLUSION Les chorées représentent une symptomatologie peu fréquente dans notre contexte. Dans notre étude la chorée de Sydenham est la forme la plus représentée des chorées aigues infectieuses de l’enfant. Le traitement par l’halopéridol garde toute son efficacité dans le traitement symptomatique des chorées.
Conflit d’intérêts : Aucun des auteurs n’a de conflit d’intérêts à signaler. Divulgation : L’étude n’a reçu aucun soutien financier ni dans sa mise en œuvre, ni pour une orientation des résultats. La seule guidance fut l’intérêt scientifique. Remerciements: Nous tenons à remercier les personnes sans lesquelles cette étude n’aurait pas été possible.
Tableau I : Répartition des patients en fonction des pourcentages des tranches d’âges.
Tableau II : tableau récapitulatif des enfants diagnostiqués de chorée de Sydenham.
CG : Chorée généralisée ; HC : hémichorée ; PA : Polyarthralgie ; IM : Insuffisance mitrale ; IAo : Insuffisance aortique ; TDM : Tomodensitométrie ; IRM : Imagerie par Résonnance Magnétique ; N : Normal ; HL+CL : Halopéridol+ Clonazepam ; F : Favorable ; R : Récidive NEUROENDOSCOPIC TRANSVENTRICULAR TREATMENTT OF CYSTIC CRANIOPHARYNGIOMA: A CASE REPORTBACKGROUND Craniopharyngiomas are rare benign epithelial tumours, originating in the pituitary stem or pituitary gland and developing in the sellar and suprasellar region. Their treatment is still subject to debate (12). Radical surgery makes sense in the face of this benign extracerebral tumour. However, this is major surgery, given the deep localisation of the tumour and its sensitive anatomical relationships with the vasculonervous structures of the base. The totally solid forms of craniopharyngioma represent only 10%, while the cystic or predominantly cystic forms can reach 60% (4,15). These cysts can become large, compressing the optic pathways and the hypothalamus and lead to obstructive hydrocephalus. In these cases, to avoid surgical morbidities, alternative surgical strategies aimed at shrinking the cyst may be used (8). The transcortical transventricular neuroendoscopic approach represents an interesting alternative for these cystic or predominantly cystic forms. In this article, we report a case of cystic craniopharyingioma urgently treated by pure transventricular endoscopic route without catheter placement. In this case, the pure transventricular neuroendoscopic approach was simple and safe with good long-term results. CASE PRESENTATION A 4-year-old girl with no pathological medical history was received in the emergency room of our hospital for progressive disturbances of consciousness. Questioning the parents revealed a history of severe headache, nausea, visual disturbances and behavioral disturbances for the past 2 months. The physical examination found a patient with disturbed consciousness (Glasgow Coma Scale 9/15) without evidence of neurological localization. Magnetic resonance imaging (MRI) of the brain showed a large cystic lesion of the sellar and suprasellar region extended under the forehead and behind, compressing and occupying the third ventricle with obstruction of the Sylvius aqueduct (Fig. 1A). This cystic lesion was hyperintense on the T2 sequences (Fig. 1A, 1B, 1C) and T1 (Fig. 1D), and obstructed the two Monroe foramens resulting in bi-ventricular hydrocephalus with significant transependymal resorption (Fig. 1B, 1C). The biological and hormonal results were normal. In the face of this radio-clinical picture, an emergency endoscopic operative decision was made. After general anesthesia, the patient was placed in supine position, with the head in neutral position slightly raised and fixed. Through a drill hole 3 cm from the midline and 1 cm anterior to coronal suture, the right frontal horn was approached by a rigid endoscope with a 30° lens. In the lateral ventricle cavity, the dome of the cyst was identified, filling the Monro foramen (Fig. 2A). A large opening of the cyst was made using monopolar coagulation and scissors, letting it escape under strong pressure; yellowish, oily liquid rich in shiny fragments (Fig. 2B, 2C and 2D). Abundant and progressive washing made it possible to obtain complete drainage of the cystic contents. Then, the endoscope was introduced into the cyst through the stoma, which allowed visualization of the yellowish deposits on the internal wall of the cyst and to drain as much as possible by rinsing (Fig. 2E and 2F). No communication between the cyst and the basal cisterns was made due to the absence of anatomical landmarks. Collapsing of the cyst and release of the Monro foramen were evident when the endoscope was removed. The closure was carried out according to the conventional technique. Drug treatment with corticosteroids was administered postoperatively. The immediate postoperative follow-up was simple with a clear improvement in clinical symptoms. There were no postoperative complications, including chemical meningitis. The postoperative control MRI performed 27 months after the operation showed the collapse of the cyst with significant reduction in its size and release of the CSF circulation pathways (Fig. 3). Note that the signal for cystic content on the control MRI was identical to that of the CSF, suggesting its continuous dilution (Fig. 3). DISCUSSION Craniopharyngioma is a benign brain tumour of embryonic origin which can be solid, cystic or mixed. For many decades and all over the world, neurosurgeons recommended the complete removal of this histologically benign tumour because it was supposed that its complete surgical removal should, in theory, allow its cure. Many surgical approaches have been used to remove these lesions, including the pterional, sub-temporal, interhemispheric, transcortical, transcallosal, transsphenoidal and translamina terminalis approaches (7,10,13,14). However, the often-intimate relationships of the tumour with delicate neurovascular structures and the frequent absence of a cleavage plan have modified this strategy towards less aggressive approaches. The search for new surgical management strategies as an alternative to the excision of craniopharyngiomas is primarily related to the high recurrence rate and the appearance of neurological sequelae incompatible with a good quality of life for some of the patients. The transsphenoidal endoscopic approach allows, in some cases, the removal of the tumour without manipulation of the brain, and has been associated with a reduction in the risk of postoperative complications (9). However, dissecting the tumour from the hypothalamus and the optic pathways, even through this minimally invasive surgical approach, may be the cause of a significant morbidity (8). The totally solid forms of craniopharyngioma represent only 10%, while the cystic or predominantly cystic forms can reach 60% (4,15). These cysts can become large, compressing the optic pathways and the hypothalamus and lead to obstructive hydrocephalus. In these cases, to avoid surgical morbidities, alternative surgical strategies aimed at shrinking the cyst may be used (8). Permanent and repeated cystic drainage can be effective even in the long term in most cases, since the clinical symptomatology of these cystic lesions is more related to the mass effect than to tumour infiltration. Conventionally, the management of these cystic craniopharyngiomas consists of placing a catheter in the cyst, connected to a subcutaneous reservoir, for the periodic aspiration of the cystic contents but also for the intracystic injection of chemo- and radiotherapeutic agents, to control tumour growth and delay the potentially harmful effects of surgery or radiotherapy (3). The use of neuroendoscopic positioning of intracystic catheters is considered to be safer than the stereotaxic approach, since the capsule of the cyst can be thick and visual control is preferable in this case. The concept of marsupialisation of the cyst in the ventricles, aimed at the continuous dilution of the cyst fluid and its resorption by the CSF pathways was introduced in 1997 by Spaziante and Divitiis (15). This cystoventriculostomy therefore represents an evolution of the first experiences of drainage of cystic craniopharyngiomas carried out by the pioneers (5,6,16). The transventricular neuroendoscopic approach is a minimally invasive option suitable for cystic craniopharyngiomas which are invasive or develop in the ventricular system. Under direct vision control, this approach, which allows total drainage of the cyst with wide marsupialisation in the ventricular system, represents obvious advantages over other drainage techniques (11). The operability of the instrument allows partial resection of the capsule for diagnostic purposes and especially for wide marsupialisation (11). During this procedure, special attention should be paid to complete elimination by aspiration and abundant rinsing of the cystic contents, since its leakage into the ventricles could possibly lead to chemical meningitis. Cysto-cisternostomy, making the cyst communicate with the basal cisterns, can be performed but remains a risky action in an area rich in delicate vascular and nervous structures and where the anatomical landmarks are not always identifiable. Some use stenting, which consists of placing a cysto-ventricular catheter to promote self-elimination of the cyst (8). Cysto-cisternostomy and stenting, which are measures to prevent reclosure and recurrence, were not performed in our case. In the context of a developing country such as ours, the high cost and unavailability of catheters and intracystic chemotherapy or radiotherapy make the endoscopic transventricular approach with a simple evacuation drainage and a wide marsupialisation of the cyst in the CSF circuit, a true minimally invasive alternative in the treatment of these cystic lesions. Even if the drainage and endoscopic transventricular marsupialization result in a significant and lasting regression of the cyst size, it remains nevertheless a temporary surgical option and requires regular clinical follow-up and close neuroradiological monitoring at 3 months, 6 months, and 12 months after surgery and then every 12 months (8). The use of intracavitary adjuvant treatments requires a sealed cavity, confirmed by a permeability test, which is incompatible with large marsupialisation of the cyst (11). In addition, the efficacy of these adjuvant treatment on prolonging recurrence-free survival in patients with cystic craniopharyngiomas is controversial and no randomised study has yet shown their superiority compared to a simple puncture-aspiration (1,2,3). In our case, the simple drainage of the cyst with a large marsupialisation in the ventricule, without cystocisternostomy, or stenting, allowed us to obtain a considerable reduction in the size of the cyst as well as its continuous dilution in the long term (27 months) with disappearance of any mass effect and consequently lasting resolution of clinical symptoms. CONCLUSION In cases of cystic or predominantly cystic craniopharyngioma, when symptoms are caused by the cystic component of the tumour, the minimally invasive transventricular neuroendoscopic approach targeting drainage and marsupialisation of the cyst may be sufficient and should be recommended. In our case, this minimally invasive approach has proven to be safe, simple and effective over the long term. However, it remains a temporary surgical option and requires regular clinical follow-up and close neuroradiological monitoring. Funding: No funding utilized from any organization. Conflicts of interest: None.
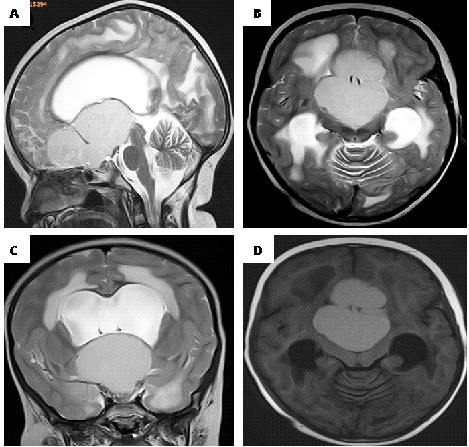 Figure 1. preoperative MRI images A-B-C: T1-weighted and D: T2-weighted showing a large cystic lesion of the sellar and suprasellar region extended under the forehead and behind, compressing and occupying the third ventricle. The cyst obstructs the two Monro foramens, causing a bi-ventricular hydrocephalus with a significant transependymal resorption.
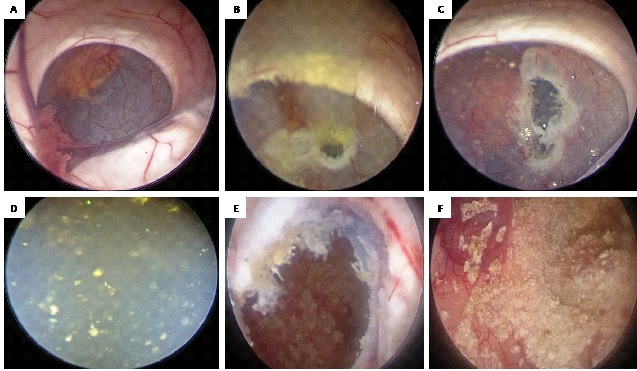 Figure 2. intraoperative neuroendoscopic images: A: view of the interior of the right lateral ventricle showing the cyst obstructing Monro foramen. B: View after fenestration of the cyst showing the oily liquid escaping under pressure. C and D: view after enlargement of the fenestration and rinsing showing the cystic liquid filled with shiny fragments. E and F: view of the interior of the cyst after complete drainage of its contents, showing yellow deposits on its internal wall.
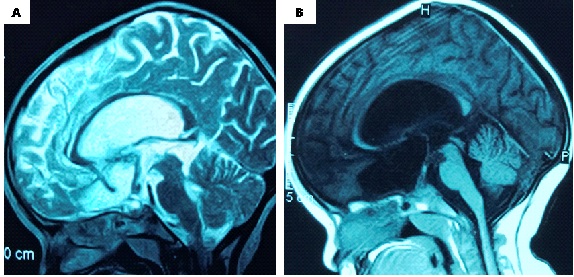 Figure 3. A: sagittal T2-weighted and B: T1-weighted post-operative MRI images (27 months) showing a considerable decrease in the size of the cyst with release of the LCS circulation pathways. Note: the sign from the cyst is identical to that of the LCS, suggesting continuous dilution. INTRODUCTION L’empyème intracrânien est une collection suppurée développée dans des espaces virtuels extradural ou sous dural (3). C’est une infection de l’encéphale due à des germes aérobies et anaérobies parmi lesquels les plus fréquents sont les streptocoques, les germes à gram négatif, les entérobactéries et les staphylocoques dorés. Ces infections parenchymateuses sont souvent secondaires à des infections de voisinage (otite, sinusite, mastoïdite) à des traumatismes crâniens ou faciaux, à la migration d’emboles septiques. Le diagnostic de cette pathologie a été rendu facile grâce au développement des techniques d’imagerie cérébrale (TDM, IRM). Elle reste une pathologie rare mais dont la mortalité et la morbidité demeurent préoccupantes (14). Le traitement d’un empyème est une urgence absolue. Le retard de prise en charge thérapeutique reste le principal facteur de mauvais pronostic (13). Ce traitement est médico-chirurgical associant une antibiothérapie à la réalisation d’une craniotomie permettant une évacuation plus complète de la collection et, ainsi, une réduction plus importante de l’inoculum bactérien (14). La tuberculose est une maladie infectieuse due au Bacille de Koch, elle est fréquemment retrouvée au niveau des poumons. Néanmoins des formes extrapulmonaires sont retrouvées ; elles représentent 15 à 30 % des cas (1) et sont souvent rencontrées chez les patients séropositifs au VIH/SIDA (5,11). Les ganglions et la plèvre constituent les localisations extrapulmonaires les plus fréquentes suivies par les formes ostéo-articulaires et uro-génitales (6,7). Les formes neuromeningées et digestives sont très rarement retrouvées (2). L’empyème extra dural d’origine tuberculeuse chez un sujet jeune immunocompétent est le 1er cas rencontré dans notre expérience, et nous permet de discuter les aspects cliniques et thérapeutiques de cette localisation. OBSERVATION Un patient mélanoderme âgé de 33 ans, sans antécédent médico-chirurgicaux particuliers avait consulté pour tuméfaction pariétale gauche évoluant depuis six (6) mois. L’interrogatoire ne retrouvait pas de notion de contage tuberculeux mais la réalisation 9 mois plus tôt d’une biopsie d’adénopathie cervicale droite dont l’analyse anatomopathologique avait conclu à une adénite d’origine tuberculeuse. Le patient n’aurait bénéficié d’aucune prise en charge. L’examen physique retrouvait un état général conservé, une apyrexie, une tuméfaction pariétale gauche d’environ 6 cm de grand diamètre avec un pertuis laissant sourdre du pus. Cette tuméfaction était non inflammatoire ferme avec absence d’écoulement du pus à la pression. Par ailleurs on notait la présence de multiples adénopathies cervicales au niveau latéral supérieur gauche abcédées, postérieur gauche en voie de cicatrisation et également au niveau sus-claviculaire gauche et droit non fistulisées (Fig 1). L’examen des autres appareils était normal. Le bilan radiologique notamment l’IRM cranio-encéphalique et cervicale en coupes axiales, sagittales et coronales en T1 et T2 avant et après injection de Gadolinum objectivait au niveau cérébral en pondération T1 un hypersignal extra axial pariétal gauche bien limité se prolongeant en extra crânien par un défect osseux (Fig 2), en pondération T2 un hyposignal entouré d’un hypersignal (Fig 3). Au niveau cervical une image hétérogène bien limitée bilatérale (Fig 4). Le bilan biologique ne trouvait pas de syndrome infectieux, la sérologie rétrovirale était négative. Le diagnostic de tuberculose extra-pulmonaire a été retenu devant les adénopathies multiples, le résultat anatomopathologique de la biopsie faite plus tôt et l’absence de traitement spécifique. Le patient a été mis sous quadrithérapie antituberculeuse puis opéré une semaine plus tard. Sous anesthésie générale, au niveau crânien nous avons fait une exérèse des tissus nécrotiques sous cutanés à l’aide de la monopolaire (Fig 5) et à travers le défect osseux, élargie à l’aide de Kérisson nous avons évacué le pus enkysté par curetage (Fig 6). Au niveau cervical nous avons fait un curage des ganglions abcédés. Les suites opératoires immédiates étaient simples. L’évolution était marquée par la disparition de l’abcès pariétal, des adénopathies cervicales et une cicatrisation des plaies (Fig 7 et 8). Nous avons réalisé un scanner crânio-encéphalique de contrôle qui est revenu normal. Le patient a été déclaré guéri et le traitement arrêté huit (8) mois plus tard. Les examens histopathologiques des différents prélèvements réalisés au bloc opératoire ont conclu à une granulomatose nécrosante encéphalique et à des adénites tuberculeuses en cervical. DISCUSSION Il s’agit d’un cas rare de tuberculose extra pulmonaire qui s’est présenté sous la forme d’un empyème extradural chez un sujet jeune immunocompétent. La tuberculose extra pulmonaire est une pathologie de plus en plus fréquente (6,7) qui a connu une résurgence avec la pandémie à VIH (5,11). La plèvre, les ganglions, les os, les articulations et les organes génito-urinaires seraient les plus touchés et très rarement le système digestif et neuroméningé (2). Au niveau neuroméningé, le bacille de Koch est souvent responsable soit d’une méningite tuberculeuse, d’u abcès intra parenchymateux, d’un tuberculome isolé ou associé à une méningite (4). Nous n’avons pas retrouvé de cas d’empyème intracrânien dans la littérature, il s’agit donc du premier cas décrit. Dans notre cas, l’hypothèse la plus probable de cette localisation serait une contamination locorégionale à partir des adénopathies cervicales par embole septique. Le BK aurait regagné la zone pariétale via l’artère carotide externe et ses terminaisons. Puis par contiguïté, il aurait infiltré l’os, créé une lyse osseuse et pénétré en intra crânien. Sur le plan clinique, ce patient n’a présenté aucun signe généralement retrouvé dans les empyèmes : fièvre, troubles de la conscience, signes de localisation, crises comitiales (12). Cela pourrait être le résultat de plusieurs facteurs mis ensemble, à savoir l’évolution lente sur plusieurs mois de la maladie, l’automédication systématique généralement observée, la localisation extra-durale et l’aspect enkysté du pus. Le retard de la mise en route du traitement de la tuberculose ganglionnaire initialement diagnostiquée chez ce patient serait dû à la survenue de la maladie lors de la pandémie à SARS COV 2. Pendant cette période, les hôpitaux étaient réquisitionnés, reléguant au second plan la prise en charge des pathologies non urgente. Avant l’avènement des antituberculeux spécifiques, la chirurgie était l’outil thérapeutique de base des formes extra pulmonaires de la tuberculose (8), il se pose ici le problème de la place de la chirurgie dans ces formes surtout avec la résurgence de cette maladie et l’émergence de la tuberculose multirésistante. La tuberculose reste cependant avant tout une maladie médicale dont le traitement, bien codifié, comporte deux phases. Une phase initiale de deux mois comprenant quatre molécules : isoniazide (4–6 mg/kg une fois par jour), rifampicine (8–12 mg/kg une fois par jour), pyrazinamide (20–30 mg/kg une fois par jour), éthambutol (15–25 mg/kg une fois par jour) et une phase d’entretien de durée variable comprenant deux molécules (isoniazide, rifampicine). La durée du traitement dépend de la localisation (9,10). Cependant le recours à la chirurgie est parfois nécessaire dans les suppurations intracrâniennes avec une collection importante pour faciliter la guérison. Pour cette forme extra-durale de la tuberculose neuroméningée, nous préconisons systématiquement un traitement chirurgical associé au traitement médical. Nous avons réalisé une craniectomie au lieu d’une craniotomie par élargissement du défect osseux existant. Ce patient a bénéficié d’une quadrithérapie antituberculeuse pendant deux mois puis d’une bithérapie six mois supplémentaires avec des contrôles scannographiques en post opératoire immédiat et à la fin du traitement antituberculeux. Le patient n’a présenté aucun effet secondaire lié au traitement. L’évolution était favorable et la guérison déclarée à la fin du huitième mois de traitement anti tuberculeux. CONCLUSION L’empyème extra dural, chez un patient jeune immunocompétent associé à des adénopathies cervicales ayant un caractère chronique doit faire penser à une tuberculose neuroméningée de localisation extra durale bien que rare. Cette étiologie sera par la suite confirmée par les examens histopathologiques. La tuberculose même dans cette forme très rare d’empyème extra dural reste une maladie médicale dont le traitement est bien codifié. Cependant le nettoyage chirurgical garde une place primordiale dans la prise en charge.
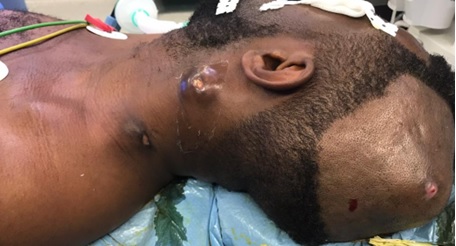 Figure 1 : Image pré opératoire
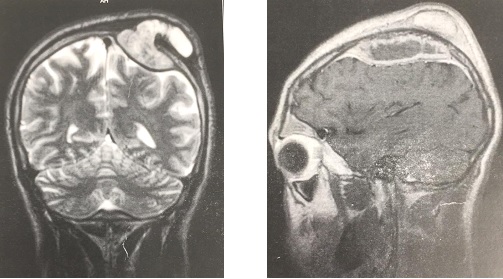 Figure 2 et 3 : IRM crânio-encéphalique, coupe coronale pondération T1 et sagittale pondération T2 :
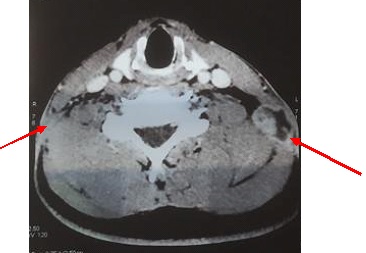 Figure 4 : IRM cervicale, coupe axiale
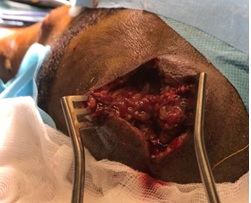 Figure 5 : Image per opératoire
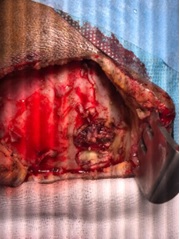 Figure 6 : Image per opératoire
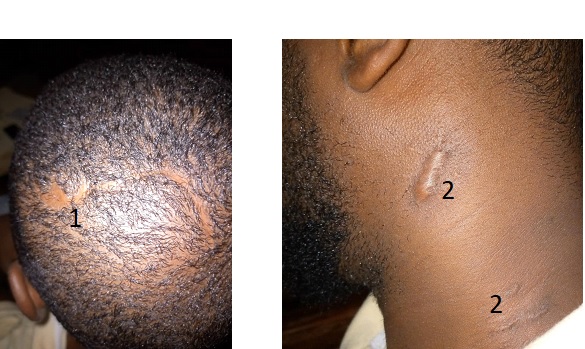 Figure 7 et 8 : Evolution à 8 mois du post opératoire
Articles récents
Commentaires récents
Archives
CatégoriesMéta |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
© 2002-2018 African Journal of Neurological Sciences.
All rights reserved. Terms of use.
Tous droits réservés. Termes d'Utilisation.
ISSN: 1992-2647