
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
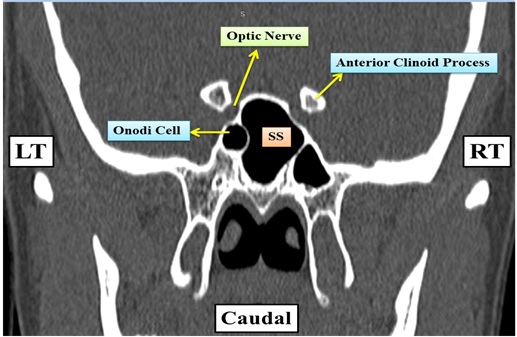
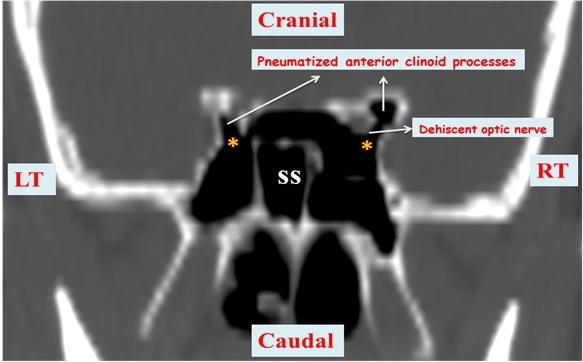
INTRODUCTION: Onodi (sphenoethmoidal) cells are described as variants of the most posterior ethmoidal cells, which pneumatises superiolateral to the sphenoid sinus. Named after the Hungarian rhinolaryngologist, Adolf Onodi, they lie intimately related to the optic canal, and may extend into the anterior clinoid processes (9,10.13,22). The endoscopic endonasal transsphenoidal approach to the base of the skull is now widely being employed in the management of diseases affecting structures in the vicinity of the sphenoid sinus, such as, optic nerve decompression and pituitary hypophysectomy. This is because, this route is quicker, safer, and a direct access to the sphenoid, with better visualisation, that allows a more complete removal of a pituitary adenoma (28). However, during this procedure, an unidentified Onodi cell, within the field of operation, or a dehiscent wall of the optic canal, increases the risk of inadvertent injury to the optic nerve (21). With the resultant complication of blindness, patient’s life could become frustrating with hopelessness. The use of computerized tomography (CT), as an investigative tool, has improved the quality and quantity of available information, derivable from the assessment of the paranasal sinuses, which are not directly visible using other radiologic procedures like the X-ray (3,32). Therefore, in order to prevent or control these complications, pre-interventional CT assessment of the sphenoid sinus anatomy, Onodi cells, if present, and its relationship with the optic nerve, is essential for a safe endoscopic transsphenoidal procedure. The prevalence of Onodi cells was 13% each, (11,24), in a study of 30 cadavers, and among 100 Sudanese subjects, although very low prevalence of 5% has been reported (12). Similarly, other studies have identified higher Onodi cell incidences; 18% in 100 subjects (18), and 30.6% among 350 Chinese subjects (17). In Nigeria, and Africa, there is a general paucity of data on the prevalence of Onodi cells using CT. As such, the aim of this study was to determine the prevalence of Onodi cells, and assess their relationships with the optic nerve by means of computerised tomography. We have also reported the prevalence of dehiscence of the optic canal and pneumatisation of the anterior clinoid process, due to their relationship to Onodi cells, when present, and their common ability to increase the chances of injury to the optic nerve during surgery. MATERIALS AND METHODS: Three hundred and twenty three adult sphenoid sinuses, obtained from individuals with age, ranging from 18 to 80 years, (mean age, 41.4 years ± 17.8), were retrospectively studied at the Radiology Department of the Usmanu Danfodiyo University Teaching Hospital, Sokoto, following institutional ethical approval. CT images with evidence of sinus disease, surgery, craniofacial anomalies or tumors distorting the normal anatomy of the skull base and sphenoethmoidal region were excluded. All images were taken between November 2014 and October 2019, using a GE Bright Speed Multidetector Helical CT (GE Healthcare, U.S.A, 2005) Scanner, at 200 mAs, 120 KVp, 15 cm Field of View, slice thickness of 2.5 mm, 512 X 512 matrix and a standard reconstruction algorithm. The CT slides were viewed on the computer using the Digital Imaging and Communication in Medicine (DICOM, Poland) viewer, powered by the RadiAnt Version 4.2 software. Onodi cells were studied and identified on axial and coronal reconstructed images, as aerated cavities on the superiolateral aspects of the body of the sphenoid sinus, either unilateral or bilateral, and noted to occasionally extend into the anterior clinoid processes (2,16) (Figure 1 and 2). Pneumatisation of the anterior clinoid process and dehiscence of the bony walls of the optic nerve were also studied on similar planes. Three dimensions (the anteroposterior (AP), craniocaudal (CC), and transverse (TR), diameters), of the sphenoid sinus were measured on sagittal reformatted, axial and coronal reconstructed CT images, while the volume was calculated, using the ellipsoid formula; Sinus Volume = 4/3 x π x A x B x C/23 (1), where, A, B, and C are the ellipsoid diameters corresponding to AP, CC, and TR diameters respectively. Data was tabulated and entered into computer using Microsoft Excel. SPSS Version 22 was used for data analysis. Statistical tests were employed for data analysis. Comparison of mean values of measured parameters in relation to sex and age distribution of the subjects were carried out using one way analysis of variance (ANOVA), while proportions were compared using chi-square test. Logistic regression was carried out to test for association between the presence of Onodi cells and age, sex, and dimensions of the sphenoid sinus. RESULTS: Prevalence of Onodi cells in the population From this study, the prevalence of Onodi cells was 4.0% (13 subjects). 0.9% (3 subjects) were unilateral, while, 3.1% (10 subjects) were bilateral (Figure 3). Prevalence of optic nerve dehiscence and pneumatisation of the anterior clinoid process The prevalence of optic nerve dehiscence from this study was 7.74% (25 subjects). This occurrence was more frequently bilateral (3.71%), than located on either right or left sides (Table 1). Pneumatisation of the anterior clinoid process occurred in 20.12% (65 subjects), and this incidence was also frequently bilateral (13.00%) (Table 1). In Figure 3, females had higher prevalence of both bilateral and overall incidence of optic nerve dehiscence and pneumatisation of the anterior clinoid process respectively. Relationship between age/sex and Onodi cells There was a statistically significant relation between the presence of Onodi cells and age of subjects (p = 0.015), (Table 2). Onodi cells were more frequently found among males (8 subjects; 2.5% ; 2 unilateral and 6 bilateral), than in females (5 subjects; 1.5%, 1 unilateral, and 4 bilateral) (Figure 3). However, there was no statistically significant relationship between the presence of Onodi cells and sex of subjects (p = 0.282) (Table 1). Logistic regression: Test of association between sinus dimensions, age and sex, and the presence of Onodi cells From this study, there was a statistically significant association between the anteroposterior diameter (length) of the sphenoid sinus and the presence of Onodi cells (Table 2). Interestingly, there was no statistically significant association between the degree of pneumatisation of the sphenoid sinus and the occurrence of Onodi cell (Table 2). DISCUSSION Onodi cells are embryologically derived from ethmoidal cells. They however, undergo differentiation to become intricately related to the anterior and superior-lateral aspects of the sphenoid sinus, the optic nerve, and internal carotid artery (29). The occurrence of optic neuropathy from pathological processes in Onodi cells is an indication of the intimacy between it and the optic nerve, as the nerve is frequently found running within the small cavity of the Onodi cell (16). Knowledge of the presence of an Onodi cell is key to prevent inadvertent injury to the optic nerve during endoscopic sinus and skull base surgeries (29,30). Notwithstanding, Onodi cells could give rise to isolated mucoceles, that consequently compress the optic nerve resulting in acute onset of visual loss (14,15,31). According to Driben (6), Onodi cells are more common anatomical variants of the sphenoid sinus than previously appreciated, and the incidence ranges from 8% to 14% (9,11,24,27,30). However, higher prevalence of 18% has been reported in Romania (18), 30.6% among Chinese subjects (17), 49.5% (13), and 65.3% (29). A prevalence of 5% was reported among Indians (12), however, among Nigerians in the southwest, Onodi cells occurred in 18.4% of subjects (7). Our study found a prevalence of 4%; this is lower than the results of these previous studies, despite the relatively higher number of our sample size. The differences may be due to the fact that, a number of these previous studies were conducted on cadavers with a relatively small number of patients (3,28), and others were carried out by endoscopic examination (25). The differences in the slide thickness, environmental, genetic and racial factors may have contributed to the differences between these studies. With the several CT protocols for identifying and defining Onodi cells (15), some studies have suggested that a combination of CT assessment and transnasal endoscopic examination would be necessary to avoid missed Onodi cells on CT (6,25). With the low prevalence of Onodi cells in this study, we think, it may be important to apply this combination of methods, to ensure that, a satisfactory and accurate prevalence for Onodi cell in the population is achieved. Pneumatisation of the anterior clinoid process and dehiscence of the optic canal are normal variations of the complex anatomy of the sphenoid sinus. Identifying them is critical, to minimize surgical risks and decrease morbidity (17,29). The pneumatisation of the anterior clinoid process could extend, such that, its edges fuse and blends with the walls of the sphenoid sinus. This would obscure the landmark it provides duces in its relationship with the optic nerve, which now become exposed within the sinus (17). In addition, dehiscence of the optic canal gives rise to bulging, such that, a whole, or part of the optic nerve becomes bare and unprotected (29). These conditions lead to optic nerve protrusion into the super-lateral aspect of the sphenoid sinus, thus, increasing its risks of inadvertent surgical injury (17). In this study, pneumatisation of the anterior clinoid process occurred in 20.12% (65 subjects); this is higher than the 10%, among 350 Chinese subjects (17), and the 9.2% in 300 Japanese patients (19). However, other studies have shown that, the prevalence of pneumatisation of the anterior clinoid process could range from 4 to 29.3% (4,5,23,26). The prevalence of optic nerve dehiscence from this study was 7.74% (25 subjects). This is higher than the 4%, in 25 dissected cadavers in Florida (8), 5%, among 200 Romanian subjects (18), 1.8%, and 1.5%, among Hispanics and African Americans respectively (29), but similar to the prevalence 7.7%, among Asians, and 7.4%, among Caucasians (29). The wide differences in the sample sizes, methodology (dissection/CT), racial, environmental and genetic factors may have contributed to the differences observed in these results. The statistically significant relationship between age and presence of Onodi cell, from this study reveals that, with advancing age, the chances of identifying an Onodi cell on CT increases. However, this was not the case with sex of subjects. CONCLUSION: The prevalence of Onodi cell among a population of north-western Nigeria was low. Increasing age of subjects and the anterioposterior diameter of the sphenoid sinus were associated with the presence of Onodi cells. An adequate pre-interventional method of identification and confirmation of the presence of Onodi cell is the key for a safe endoscopic sinus surgery. Acknowledgment: We acknowledge the department of Radiology, Usmanu Danfodiyo University Sokoto. Conflict of interest: The authors declare no conflicts of interest
Table 1. Prevalence and location of optic nerve dehiscence and pneumatisation of the anterior clinoid process
Table 2: Test of association between the presence of Onodi cells and features of the sphenoid sinus
Figure 1: Coronal CT of sphenoid sinus of a 68 year old male showing a left sided Onodi cell and its relation to the left optic nerve. SS = Spenoid Sinus cavity, RT = Right Side, LT = Left Si
Figure 2: Coronal CT of sphenoid sinus of a 52 year old male showing bilateral Onodi cells (marked ⃰), a dehiscent right optic nerve, and bilaterally pneumatised anterior clinoid processes just lateral to the optic canal. SS = Spenoid Sinus cavity, RT = Right Side, LT = Left Si
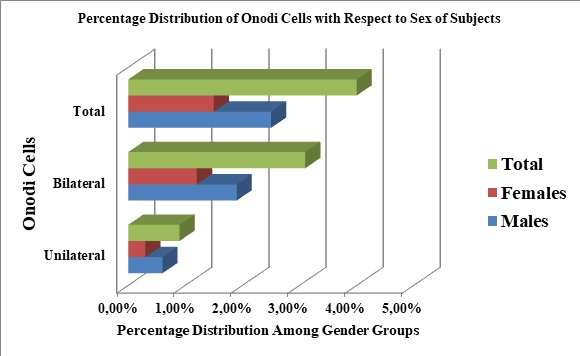 Figure 3: Prevalence of Onodi cells in the studied population
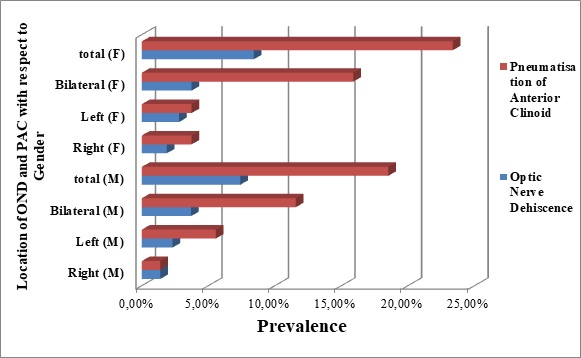 Figure 4: A 3-D bar chart, showing the locations (sides) and prevalence of optic nerve dehiscence (OND), and pneumatisation of the anterior clinoid process (PAC), among the genders (male = M, female = F). INTRODUCTION La thiamine (ou vitamine B1) est une vitamine hydrosoluble qui passe la barrière hémato-encéphalique, d’origine uniquement alimentaire (pas de synthèse endogène de la thiamine), elle est présente dans presque toutes les viandes (volaille, porc…), céréales (riz, son…), légumes et légumineuses. Elle est absorbée par le duodénum et est stockée principalement dans le foie mais également au niveau du cœur et des muscles. Les besoins journaliers sont de 1,1 à 1,2 mg/j, et en cas de déficit, les troubles apparaissent au bout de 2 à 3 semaines. La thiamine est la forme active de la thiamine pyrophosphate ou thiamine diphosphate, elle représente un cofacteur des pyruvates déshydrogénase, de l’acide alpha-ketoglutarique, et de la trans-ketolase ; ce sont trois enzymes clés du métabolisme des carbohydrates. Le pyruvate déshydrogénase catalyse la conversion du pyruvate à l’acétyl-CoA. Ce métabolisme nécessite la présence de la thiamine pour empêcher l’accumulation des lactates (7). Le cycle de l’acide citrique est une voie métabolique centrale impliquée dans la régulation des glucides, des lipides et le métabolisme des acides aminés et sa carence ou son déficit complet, génère une accumulation de l’acide lactique, et produit une acidose lactique très sévère en inhibant la production de nombreuses molécules, y compris les neurotransmetteurs acide glutamique et GABA. La thiamine peut également participer directement à la neuromodulation (14,23). La thiamine dans le corps humain a une demi-vie de 18 jours et est rapidement épuisée, en particulier lorsque la demande métabolique dépasse l’apport. Le déficit en thiamine peut être en rapport avec une alimentation inadéquate, une baisse de l’absorption, un défaut d’utilisation, ou un excès d’utilisation, il peut survenir chez les patients sévèrement atteints, en cas d’augmentation du métabolisme du glucose (ex. choc septique, postopératoire…), en cas de malnutrition prolongée (ex : syndrome de re-nutrition) ou en cas d’une élimination excessive (ex : atteinte rénale sévère). Dans tous les cas, une supplémentation parentérale ou entérale est primordiale pour éviter les acidoses lactiques secondaires à la carence en thiamine. L’encéphalopathie de Wernicke a été décrite pour la première fois en 1881 par Carl Wernicke, avec la triade classique : paralysies oculomotrices, troubles de la conscience et ataxie. Il s’agit d’une complication neuropsychiatrique aiguë secondaire à un déficit en thiamine, fréquemment rencontrée chez les grands consommateurs d’alcool. Dans le cas d’un traitement inadéquat, le déficit en thiamine provoque des lésions structurelles cérébrales définitives, qui se manifestent par des troubles mnésiques antérogrades et rétrogrades sévères et souvent irréversibles (25). Actuellement, différents tests mesurant l’activité de la thiamine pyrophosphate (forme active de la thiamine) ont été développés. Ils permettent d’identifier les patients carencés qui sont à risque de développer une encéphalopathie de Wernicke (EW). Ces tests ne sont cependant pas disponibles dans l’urgence. Le diagnostic d’EW doit être posé de manière présomptive afin de traiter les patients aussi rapidement que possible (4). CAS CLINIQUE Mme M.L., âgée de 27ans, sans antécédents médico-chirurgicaux ni alcoolisme notables, suivie depuis Août 2014 pour une leucémie aigue promyelocytaire (LAM3), avec au caryotype une trisomie du chromosome 8 (15,17) et un gène FLT3 muté. Elle a été initialement traitée selon le protocole APL 2006. L’induction a été compliquée d’une pancréatite stade B, nécessitant l’arrêt du Vesanoid avec un relais par l’Arsenic. Ayant été réfractaire à cette première ligne de chimiothérapie, un rattrapage par Idarubicine et Trisenox a permis l’obtention d’une première rémission complète moléculaire 3 mois après le début du traitement, consolidé par 3 cycles d’Arsenic, puis un entretien par Purinethol 90mg/m²/semaine associé au méthotrexate15mg/m² /semaine par voie orale. Une rechute moléculaire a été observée 2 mois après le début du traitement d’entretien. Par la suite, elle a reçu une seconde chimiothérapie de rattrapage par Gemtuzumab-Ozogamycine (Mylotarg) associé au trioxide d’Arsenic, suivie d’une allogreffe géno-identique en 2ème rémission complète. La patiente a été conditionnée par Cyclophosphamide, associé au Busulfan. Le processus a été compliqué d’une GVH (réaction du greffon contre l’hôte) digestive grade 2 cortico-sensible. Une deuxième rechute moléculaire a été observée à 5 mois de l’allogreffe, nécessitant l’arrêt de la ciclosporine et des corticoïdes, et une perfusion de DLI (Donnor Lymphocytes Infusion). La patiente a rechuté en neuro-méningé, et a donc reçu une nouvelle induction par Gemtuzumab Ozogamycine (Mylotarg) associé au trioxide d’Arsenic et à 12 ponctions lombaires thérapeutiques. Le traitement de rattrapage s’était compliqué d’une aplasie profonde prolongée, dans un contexte de vomissements incoercibles rebelles aux traitements usuels, suite auxquels elle avait été mise sous nutrition parentérale exclusive et prolongée pendant huit semaines, sans supplémentation vitaminique. A la 6ème semaine d’hospitalisation, la patiente a présenté de façon brutale un trouble oculomoteur, à type de nystagmus vertical bilatéral, avec flou visuel bilatéral, associé à un syndrome cérébelleux cinétique qui s’est manifesté par un élargissement de la surface de sustentation, une marche avec les bras écartés, une hypermétrie, une dyschronométrie, avec des réflexes rotuliens pendulaires bilatéraux à l’examen clinique, sans déficit sensitif ni moteur. Une TDM cérébrale réalisée est revenue normale. Une IRM cérébrale faite a mis en évidence une lésion bithalamique ventrale et postérieure en hypersignal FLAIR et en hypersignal diffusion b1000 sans restriction sur la cartographie de l’ADC, sans prise de contraste après injection de Gadolinium. Figure (1). Un électroencéphalogramme a retrouvé un tracé d’organisation préservée, fait de rythme alpha postérieur moyennement volté, légèrement irrégulier, avec absence d’anomalie comitiale. Un dosage de la vitamine B1 érythrocytaire a été réalisé, en faveur d’une carence profonde en thiamine (taux de thiamine total à 79 mmol/l, pour un intervalle de normalité entre 126 et 250 mmol/l). La patiente a été supplémentée en thiamine à la posologie de 1 g/jour en intraveineux, pendant 10 jours, avec un relais par voie orale. Une IRM de contrôle faite après 3 mois a montré une nette régression de l’hypersignal bi thalamique ; une normalisation de la diffusion, une stabilité des hypersignaux FLAIR micronodulaires intéressant la substance blanche supra-tentorielle, aspécifique (Figure 2). La patiente s’est améliorée sur le plan neurologique dès les premières 48h. L’ataxie a complètement régressé dans les mois qui suivent, mais un nystagmus séquellaire persiste jusqu’à ce jour (trois ans après). REVUE DE LA LITTERATURE Les carences vitaminiques, essentiellement ceux du groupe B ; sont souvent responsables de troubles neurologiques et d’états confusionnels. Ces carences sont majoritairement associées à l’alcoolisme (1). Le déficit en thiamine peut s’observer chez des patients non alcooliques, dans la population pédiatrique et les adolescents, en cas de malnutrition, d’alimentation parentérale prolongée, de maladie gastro-intestinale, de diarrhée prolongée, vomissements récurrents, chirurgie bariatrique, hépatopathie, pathologie maligne, vomissements gravidiques et hémodialyse (19,13,18). Sur le plan anatomopathologique, l’EW est caractérisée par des suffusions hémorragiques associées à une prolifération gliale et à une démyélinisation au niveau des structures entourant le troisième ventricule, les corps mamillaires et les noyaux oculomoteurs. Des études de prévalence basées sur des autopsies montrent que l’EW est une maladie fréquente qui n’est souvent décelée qu’après le décès. A l’autopsie, on trouve des lésions caractéristiques chez environ 1,5% de la population générale et la prévalence augmente à 12,5% chez les patients alcoolo-dépendants (4,10,11) alors que 5 à 14% seulement des personnes atteintes sont identifiées avant leur décès (24). Les difficultés diagnostiques sont dues au fait que seulement 10% des patients atteints présentent simultanément la triade classique associant confusion, ataxie et ophtalmoplégie (21). D’autres signes cliniques comme des troubles cognitifs, une somnolence, un syndrome de Korsakoff (syndrome amnésique lié à des signes frontaux), un état stuporeux voire un coma, qui apparaissent chez 80% des patients présentant une EW, peuvent également être attribués à une intoxication alcoolique aiguë, à un syndrome de sevrage ou à un problème médical autre. L’ataxie est présente chez seulement 23% des patients et l’ophtalmoplégie chez 29%, d’autres symptômes tels qu’une hypotension ou hypothermie inexpliquée peuvent être présents (17,26). L’EW se manifeste donc souvent par un état confusionnel aspécifique entraînant chez 90% des patients le risque que le diagnostic soit manqué (21). Selon les recommandations de l’EFNS (European Federation of Neurological Societies), un dosage de thiamine doit être réalisé dès la suspicion clinique de l’EW, et une IRM doit être faite pour conforter le diagnostic (5). L’IRM montre une hyperdensité symétrique au niveau des corps mamillaires, du thalamus, de la matière grise périaqueducale, et des colliculi supérieur et inférieur (structure sous-corticale bilatérale située sur le toit du mésencéphale), cette hyperdensité est observée en T2 et en FLAIR (Fluid-Attenuated Inversion Recovery) (1). Le pronostic des patients insuffisamment ou non traités est sombre. Victor et al. ont observé une récupération complète chez seulement 16% des patients, avec une EW traitée avec des faibles doses parentérales de 50 à 100 mg de thiamine par jour, le taux de mortalité étant de 17 à 20% (26). Pour améliorer le pronostic des patients chez qui on diagnostique ou suspecte une EW, la thiamine doit être administrée aussitôt que possible à un dosage adéquat, et tout retard de la prise en charge augmente le taux de mortalité (3). DISCUSSION La thiamine est une vitamine exogène hydrosoluble, qui peut être stockée par le corps. C’est un cofacteur des enzymes de la voix d’oxydation des pyruvates. La carence en cette vitamine se manifeste cliniquement par une encéphalopathie de Wernicke, le diagnostic est clinique, ainsi un dosage normal en thiamine ne devrait pas retarder la supplémentation vitaminique immédiate. La triade caractéristique de l’EW était absente chez notre patiente comme dans 80% à 90% des cas décrits dans la littérature (12,15). Les troubles oculaires étaient au premier plan, avec la baisse de l’acuité visuelle, et une dysfonction oculomotrice, habituellement retrouvés chez 29% des patients atteints d’encéphalopathie de Wernicke (20,8). Les patients sous nutrition parentérale prolongée, nécessitent un apport plus important en thiamine afin de métaboliser leurs hydrates de carbone (2). Toutes les publications, mise à part une seule, ont conclu à ce que l’alimentation parentérale prolongée soit un facteur de risque primaire de l’encéphalopathie de Wernicke dans le contexte d’allogreffe (3). Notre patiente a reçu 6 semaines de nutrition parentérale exclusive en post allo-greffe, sans supplémentation en thiamine, avec apparition des symptômes à J42. Plusieurs auteurs préconisent une supplémentation en thiamine systématique en prophylaxie de l’EW, quoiqu’une publication d’un groupe brésilien, a mis en évidence le décès de 8 patients atteints d’EW, malgré une prophylaxie préalable par la thiamine à raison de 50mg par jour (3). Jusqu’à présent il n’y a toujours pas de consensus pour traiter l’EW, des essais randomisés et contrôlés doivent être menés afin de trouver la posologie et la durée optimale du traitement par thiamine, autant en curatif qu’en préventif. L’EFNS recommande 200 mg par voie intraveineuse 3 fois par jour, jusqu’à ce qu’il y ait stabilité des signes cliniques. Elle préconise également un suivi du dosage de la thiamine pendant au moins 6 mois (6). D’autre part, le NICE (National Institue for Health and Care Excellence) recommande une posologie de 500 mg ou 750 mg, 3 fois par jour pendant 5 jours. Une supplémentation par voie orale est recommandée après l’administration intraveineuse (6). Une amélioration clinique a été rapidement notée chez notre patiente dès l’initiation de la supplémentation en thiamine. L’ophtalmoplégie s’améliore en général dans les heures qui suivent la supplémentation, l’ataxie se résout en quelques jours, et la confusion mentale prend environ 3 à 4 semaines avant de s’arranger (9). Notre patiente a gardé un nystagmus séquellaire, seulement 20% des patients récupèrent complètement (3). Ce cas clinique met le point sur le rôle vital de la supplémentation en thiamine, chez les patients recevant une alimentation parentérale prolongée, surtout dans un contexte de néoplasie et de greffe. Les patients ayant subi une allogreffe sont à haut risque de développer une encéphalopathie de Wernicke, à cause de la malnutrition chronique, des nausées/vomissements chimio-induits, et de la forte consommation de thiamine par les cellules tumorales en croissance (16). CONCLUSION En conclusion, devant une symptomatologie neurologique et ophtalmologique, dans un contexte de dénutrition, de chimiothérapie lourde, de nutrition parentérale prolongée, de vomissements incoercibles, de chirurgie bariatrique, de néphropathie ou d’hépatopathie, une IRM cérébrale associée à un dosage de la vitamine B1, doivent être réalisés immédiatement. La supplémentation en thiamine devrait être débutée aussitôt que le diagnostic d’encéphalopathie de Wernicke est suspecté afin de prévenir la mortalité et la morbidité.
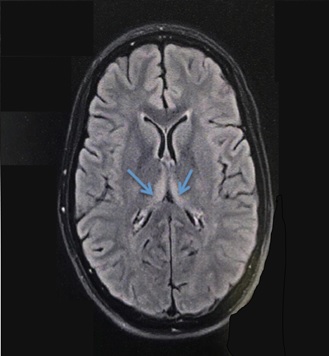 Figure 1 : IRM cérébrale montrant une lésion bithalamique ventrale et postérieure en hypersignal FLAIR. Cet aspect est évocateur d’une encéphalopathie de Wernicke
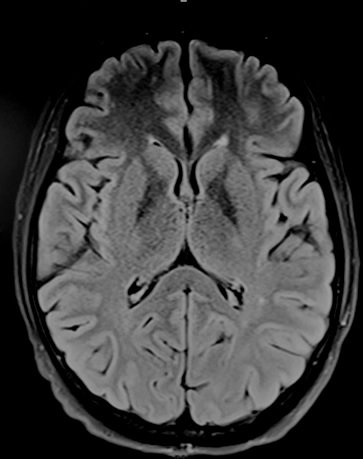 Figure 2 : IRM cérébrale, après 3 mois de supplémentation vitaminique, montrant une nette diminution de l’hypersignal bithalamique. Mesdames et messieurs, Chers confrères, Je voudrais rendre un hommage à un homme exceptionnel en la personne de Mr le Prof Vincent Ba Zeze (1952-2020) qui a été arraché à notre affection le 14 novembre 2020. Entré à l’école de médecine d’Abidjan en 1972, il fut major du concours d’internat de médecine en 1978 et débuta son internat de chirurgie générale au CHU de Treichville. En 1982, il est nommé Assistant Chef de Clinique en chirurgie option orthopédie. C’est alors que son maître Monsieur le Prof Gnangni Angate, dans une grande vision l’a orienté vers la neurochirurgie. A l’époque, il a reçu cette information comme une punition parce qu’il s’agissait d’une spécialité imaginaire en ces temps-là en Côte d’Ivoire. C’est ainsi qu’il posa ses valises à Tours de 1983 à 1988 où il intégra le service de neurochirurgie dirigé par Monsieur le Prof Michel Jan qui en a fait un excellent neurochirurgien. De retour à Abidjan en 1988, il est nommé par voie de concours Maître de conférences agrégé faisant de lui le 1er Professeur de neurochirurgie de Côte d’Ivoire, puis quelques années plus tard, Professeur titulaire. Le début d’une grande carrière pouvait alors commencer. Les challenges étaient importants mais pas assez grands pour lui, lui pour qui rien n’était impossible. Il devait se battre pour exister, faire connaitre la spécialité, opérer, au départ des hydrocéphalies, des fractures de l’étage antérieur de la base du crane, des hernies discales dans un environnement difficile, parfois hostile où il n’y avait aucun scanner, aucun service, aucun neuro anesthésiste et très peu de médecins anesthésistes. Il disait lui-même qu’il devait opérer après des péritonites, des occlusions et faire lui-même des sacco-radiculographies. Puis arriva 1990, année d’ouverture du service de neurochirurgie de Yopougon et de l’arrivée du 1er scanner. En ces temps-là ils n’étaient qu’une poignée de neurochirurgiens ; avec lui, Prof Gilbert Dechambenoit, Prof Varlet et Prof Jacques Santini qui a dirigé le service jusqu’en 1994, année à laquelle le Prof Ba Zézé lui succéda jusqu’à sa retraite. C’était un chirurgien hors pair, formé à la chirurgie crânienne et vasculaire ; il était polyvalent. Il pouvait passer avec une aisance déconcertante d’un abord rétro-sigmoïdien à un abord supra cérébelleux infratentoriel, en passant par une voie sous temporale. A cela il faut ajouter les arthrodèses cervicales et dorsolombaires. Il a opéré, opéré et encore opéré sans jamais s’arrêter, tant la demande était forte. Mais la tâche n’était pas assez grande, il lui fallait constituer une équipe en formant des neurochirurgiens, des neuroanesthesistes, des instrumentistes, des infirmiers, des aides-soignantes. Pour cela il a créé des partenariats avec des équipes de neurochirurgie, des hôpitaux, des collègues et des amis en Afrique, en Europe (France, Belgique) et aux USA. Ainsi au soir de sa retraite il avait formé 7 maîtres de conférences agrégé en Côte d’Ivoire ainsi que de nombreux neurochirurgiens en Côte d’Ivoire et en Afrique francophone. Il rêvait d’une neurochirurgie africaine dynamique et fleurissante. Toutes ses qualités lui ont valu des distinctions africaines et faisaient qu’il était apprécié de ses pairs. Par ailleurs sa famille était tout pour lui et lui était tout pour sa famille, véritable Papa gâteau. Malgré tout cela il avait une vie associative chrétienne pleine, engagée toujours au service des autres et lui-même disait : « les autres d’abord » Alors que retenir de lui : précurseur, initiateur, promoteur, formateur, simplicité, humilité, joie de vivre, spiritualité, professionnalisme, au service des autres. Maître, vous avez marché en laissant des traces, la tâche est accomplie. Merci maître !
Landry DROGBA Neurochirurgien Maitre de conférences agrégé UFRSMA-Abidjan Elève du Professeur Vincent BAZEZE
STROKE TREATMENT IN A LOW RESOURCE SETTING: THE MOTEBANG HOSPITAL PROTOCOL BACKGROUND Stroke, the largest source of long-term neurologic disability in high-income countries, is increasingly recognized as a major cause of death and disability in low- and middle-income countries as well. While stroke incidence has been decreasing over time in high-income countries, it is level or likely increasing in low- and middle-income countries to the point of exceeding the high-income country incidence(8). Stroke incidence, prevalence, and mortality appear to be particularly high in sub-Saharan Africa (32,33,39). Stroke prevention and treatment in sub-Saharan Africa is complicated by multiple factors associated with limitations of resources and community awareness, such as more limited control of hypertension, late presentation to the hospital, and unavailability of the full set of diagnostic and therapeutic modalities available in high-income countries (1,10,16,27,4,36). There also appears to be differences in epidemiology of stroke subtypes such as a higher proportion of haemorrhagic relative to ischaemic strokes and disproportionate incidence in younger individuals (age <50) (22,35), potentially attributable to the high prevalence of untreated hypertension. Lesotho, a small landlocked country in southern Africa, is challenged with the world’s second highest HIV prevalence rate (38) and has only 0.068 doctors per 1000 people (13), representing approximately 160 doctors serving a population of two million. No systematic studies of stroke epidemiology have been conducted in Lesotho, but based on studies from neighbouring South Africa (18), stroke incidence and mortality are likely to be high. Motebang Hospital is the northern region’s referral hospital and the district hospital for the Leribe district. Motebang represents a typical district hospital for the region and includes inpatient, surgical, and outpatient services. Limited available resources include X-ray, ultrasound, basic laboratory services, and auxiliary services such as physiotherapy. Stroke patients from throughout the district are referred for care at Motebang, and all referrals for tertiary services in the northern region are made from this hospital. Motebang is developing into a teaching hospital as the central training site for the country’s only post graduate program, the Lesotho Boston Health Alliance (LeBoHA) Family Medicine Specialty Training Program (FMSTP), and is a core clinical rotation site in the new Lesotho medical internship program. Evidence-based guidelines for improving outcome and secondary prevention have been developed by major international stroke organizations (2,12,15,19,21,26,37,40). These guidelines are generally not suitable for direct adoption (i.e. use as-is) into low-resource settings such as the Motebang Hospital and instead require either contextualisation (i.e. specification of additional local details for use) or adaptation (i.e. reformulation to meet local conditions) (7). We therefore convened meetings of a stroke specialist with leadership of the Motebang Hospital and LeBoHA with the goals of systematically reviewing the major international stroke guidelines and formulating an evidence-based protocol for use in the local context. The protocol described here contextualises and adapts the existing guidelines to develop protocols for emergency treatment over the first 24 hours, subsequent inpatient treatment, and secondary stroke prevention under conditions of having no stroke specialist, no access to head CT or MRI neuroimaging (and thus limited ability to differentiate between ischaemic versus haemorrhagic stroke types), and limited available treatment modalities. Although the protocol was designed for application at the Motebang Hospital, the approach can be further contextualised to similar district hospitals in low resource settings. METHODS FOR GUIDELINE DEVELOPMENT Evidence-based stroke guidelines from the American Heart Association/American Stroke Association, European Stroke Organization, the British National Institute for Health and Care Excellence, and the World Health Organization (2,11,12,15,19,21,26,37,40) were assembled by a stroke specialist (SMG) with experience in leadership of guideline development (12,19). These guidelines, listing specific recommendations for emergency treatment, early hospital care, post-stroke recovery, and secondary stroke prevention for ischaemic and haemorrhagic strokes considered separately, were then presented iteratively at a series of conferences with the Leribe District Medical Officer and LeBoHA FMSTP Senior Registrar (TN), LeBoHA FMSTP Senior Registrar (MC), LeBoHA FMSTP Director and Lesotho Ministry of Health Family Medicine Consultant (SM), LeBoHA FMSTP faculty member and Lesotho Ministry of Health Family Medicine Consultant (BB), and Volunteer Coordinator for LeBoHa and Boston University Family Medicine faculty member (AG). The conferences were held at the Motebang Hospital 2-6 September 2019. The published international guidelines were systematically reviewed to determine which recommendations could be contextualized for use at the Motebang Hospital and which required adaptation. Because of the unavailability of diagnostic neuroimaging modalities for differentiating ischaemic from haemorrhagic stroke such as CT or MRI scan at the Motebang Hospital and most other hospitals in Lesotho, the adaptation process drew on the literature analysing clinical features whose presence or absence help differentiate between these two major types of stroke (4,20,23,25,30). Other literature and sources of information reviewed during the conferences were published studies analysing stroke care and decision-making in low-resource environments (3,14,24,27), regional standard treatment guidelines and essential medication lists from Lesotho and South Africa (17,28,29). The proposed guidelines reported here reflect the consensus of the conference attendees after iterative review and discussion. PROPOSED GUIDELINES Recommended acute stroke care varies substantially between ischaemic and haemorrhagic stroke (2, 12, 21, 26, 37). However, CT scanning, the primary modality for differentiating ischaemic from haemorrhagic stroke types, is unavailable at the Motebang Hospital and most other hospitals in Lesotho. The Motebang Hospital guidelines for stroke treatment therefore focused on 1) clinical features for differentiating probable ischaemic and haemorrhagic stroke and 2) formulating additional sets of guidelines for individuals with uncertain ischaemic versus haemorrhagic stroke aetiology. Analyses of clinical features for differentiating stroke type have demonstrated a high degree of overlap between ischaemic and haemorrhagic stroke across multiple populations and relatively few clinical features able to diagnose type with high specificity (4,20,23,25,30). Clinical features most consistently overrepresented in haemorrhagic stroke are headache at onset, vomiting, and impaired level of consciousness (Table 1). These features appear to carry relatively high specificity for haemorrhagic stroke but low sensitivity, as they are absent in the majority. The clinical features most consistently overrepresented in ischaemic stroke are history of transient ischaemic attacks (TIAs, operationally defined as prior events of neurologic deficit lasting less than 24 hours), atrial fibrillation, cardiomyopathy, valvular heart disease, and systemic atheromatous disease such as coronary artery or peripheral arterial disease. Table 1 lists additional clinical features associated with stroke subtype with less consistency across studies: gradual progression of symptoms over time (versus maximal severity at onset), and extensor plantar responses (also known as Babinski signs) favouring haemorrhagic strokes; pure face/arm/leg motor hemiparesis or pure face/arm/leg sensory loss appearing in a subset of ischaemic strokes. The Motebang Hospital guidelines call on physicians initially evaluating patients presenting to the Emergency (Casualty) Unit to apply the above features to classify individuals with acute onset of neurologic deficits consistent with stroke into categories of Probable Ischaemic, Probable Haemorrhagic, and Uncertain Ischaemic/Haemorrhagic. Initial history and neurologic examination are focused on ascertaining the cardinal features of ischaemic or haemorrhagic stroke (Table 1). The Uncertain Ischaemic/Haemorrhagic category is applied when the features with highest specificity for stroke subtype are absent or are present in mixed ischaemic and haemorrhagic combinations. Guidelines for emergency care for these three stroke categories are shown in Table 2. Initial treatment for all individuals is to assure cardiopulmonary stability and apply cardiopulmonary resuscitation steps (chest compressions, airway, and breathing or C-A-B) as needed (9). Other emergency steps shared across all stroke subtypes (2,12,21,26) are 1) treat hypoglycaemia (blood glucose <3.3 mmol/l) or hyperglycaemia (blood glucose >10.0 mmol/l) to aim for a conservative target glucose in the 7.8-10.0 mmol/l range; 2) identify and treat sources of hyperthermia (temperature >38°C) and administer antipyretic medications to maintain normothermia; and 3) administer maintenance fluids with a focus on avoiding severe hyponatremia (serum sodium <130 mmol/L). The key differences in emergency treatment for the three stroke categories are use of antithrombotic therapy and blood pressure targets. For Probable Ischaemic Stroke, recommendations are to initiate antiplatelet treatment acutely with aspirin 300mg (per oral, nasogastric, or rectal administration) and acutely lower blood pressures only if ≥220/120 mm Hg or when a hypertensive emergency is present (2,21,26). Use of intravenous thrombolytics such as tissue plasminogen activator was not recommended because of the practical inability to obtain CT scan within 4.5 hours of symptom onset and the high risk of exacerbating intracerebral haemorrhage (ICH) if present. When treating blood pressures ≥220/120 mm Hg, it is recommended to aim for modest ~15% reductions over the first 24 hours using agents available at the Motebang Hospital such as nifedipine or hydralazine. For Probable Haemorrhagic Stroke, the recommendations are to avoid early antithrombotic treatment and maintain blood pressure <140/90 mm Hg with the goal of reducing risk of early hematoma expansion (2,12,37). For individuals in the Uncertain Ischaemic/Haemorrhagic Stroke category expected to comprise the majority of acute stroke patients, the recommendations are to defer acute antiplatelet therapy during the first 24 hours following symptom onset and to lower blood pressures ≥180/105 mm Hg. Guidelines for subsequent Inpatient (Ward) care are similarly categorized according to the three stroke categories (Table 3). For Probable Ischaemic or Uncertain Ischaemic/Haemorrhagic Stroke, the recommended steps include continuation or initiation of antiplatelet therapy with aspirin 75mg per day, initiation of a statin medication, and lowering of the blood pressure to <140/90 mm Hg 2 to 4 days after stability of symptoms. For TIAs or minor probable ischaemic strokes (defined as a score of ≤3 on the National Institutes of Health Stroke Scale, https://www.mdcalc.com/nih-stroke-scale-score-nihss), it is reasonable to treat with dual antiplatelet therapy of aspirin 75mg plus clopidogrel 75mg daily for a total of 21 to 30 days to reduce the risk of early recurrent stroke, followed by conversion to antiplatelet monotherapy (26). A blood pressure target of <140/90mm Hg is maintained and antiplatelet therapy is withheld for Probable Haemorrhagic Stroke. All other recommended steps are the same for the three stroke categories: initiation of prophylaxis for deep venous thrombosis after 24 hours if the patient is not walking (with subcutaneous enoxaparin and pneumatic compression stockings if available), screening for dysphagia within 24 hours and prior to oral intake, placement of a nasogastric tube if unable to swallow and early initiation of feeding, removal of urinary catheters and substitution of incontinence padding as needed to reduce risk of urinary tract infections, regular skin assessments and turning of bedbound patients to prevent pressure sores, and initiation of physiotherapy with training of family members for post-discharge therapy as soon as feasible (2,40). The remaining set of Motebang Hospital guidelines apply to secondary stroke prevention (Table 4). Recommended steps for secondary prevention following Probable Ischaemic or Uncertain Ischaemic/Haemorrhagic Stroke are long-term blood pressure control to a target of <140/90 mm Hg, aspirin 75mg daily, statin therapy, testing and treatment for diabetes mellitus, and lifestyle modifications as applicable including smoking cessation, increased physical activity, weight loss if obese, and dietary emphasis on higher consumption of vegetables, fruits, and whole grains and reduced salt, sweets, and red meat (15). If atrial fibrillation is detected during hospitalization and the patient is considered a candidate for regular monitoring of prothrombin time, it is reasonable to consider obtaining an outpatient head CT scan within 30 days of the stroke to exclude ICH and then instituting long-term anticoagulation with warfarin to a target International Normalized Ratio of 2.0-3.0. Secondary prevention steps for Probable Haemorrhagic Stroke differ in targeting blood pressure treatment to a systolic pressure <130 mm Hg (12) and avoidance of antiplatelet, anticoagulation, or statin therapy unless for another clear indication. Recommendations for diabetes control and lifestyle modifications are the same post-ICH as post-ischaemic or uncertain strokes. DISCUSSION The protocols proposed here for emergency, inpatient, and post-discharge stroke treatment represent application of the guidelines from major international stroke organizations to the practice environment at the Motebang Hospital and similar district hospitals in Lesotho. They are designed to improve stroke outcome and reduce risk of recurrence without requiring diagnostic testing or treatment modalities unavailable in this setting. They are also designed to be applied by primary care physicians and other health care practitioners without specialized training in neurology. The proposed diagnostic categories of Probable Ischaemic, Probable Haemorrhagic, and Uncertain Ischaemic/Haemorrhagic Stroke (Table 1) are defined by features of the medical history and clinical presentation that are straightforward to elicit from rapid patient and family interview and examination. Designing guidelines for the common scenario of Uncertain Ischaemic/Haemorrhagic Stroke is challenging, as goals for thrombosis and to a lesser extent blood pressure control are at cross purposes for ischaemic and haemorrhagic strokes. The Motebang Hospital guidelines seek to maximize good outcome in this situation by 1) deferring antithrombotic treatments such as aspirin until 24 hours post symptom onset, and 2) allowing acute blood pressures to range up to 180/105 mm Hg (Table 2). Initiation of aspirin within 48 hours of ischaemic stroke onset reduces post-stroke death or dependency with a number needed to treat of 79 (31). A decision analysis model suggested that acute aspirin treatment might also be effective for uncertain ischaemic/haemorrhagic strokes (3). This model was based on a (possibly optimistic) assumption of reduced post-ICH in-hospital mortality with aspirin use (risk ratio 0.886) and was sensitive to even modest increases in post-ICH mortality (risk ratio >1.057 favoured avoiding aspirin). The Motebang protocol therefore recommended initiating aspirin only after the first 24 hours, during which hematoma expansion is common (6). The <180/105 mm Hg blood pressure threshold was selected as acceptable both as an upper limit for acute ICH (12) and for avoiding hypoperfusion following acute ischaemic stroke (2,26). Despite the unavailability of many of the neuroimaging and treatment modalities incorporated by international stroke guidelines such as acute CT scanning, thrombolysis, thrombectomy, and carotid revascularization (2,12,15,21,26,37), the Motebang Hospital guidelines nonetheless offer the opportunity for substantial improvements in stroke outcome and recurrence. Steps such as dysphagia screening, use of pneumatic compression stockings, skin assessment, removal of urinary catheters, outpatient blood pressure control, and aspirin and statin therapy following ischaemic stroke are endorsed at the highest strength of recommendation by the American Heart Association/American Stroke Association (12,26,40), indicating benefit that greatly exceeds risk. An analysis of the Muhimbili National Hospital, a large referral hospital in Dar es Salaam, Tanzania, found that some recommended post-ischaemic stroke treatments with low numbers-needed-to-treat to improve outcome such as aspirin and statin use were applied to a majority of stroke patients, whereas others such as deep vein thrombosis prophylaxis were uncommonly used (27). Anticoagulation for secondary prevention of atrial fibrillation-related stroke poses substantial challenges related to the need to exclude haemorrhagic stroke and to monitor warfarin dosing (5), but is another highly effective therapeutic option. Non-vitamin K antagonist oral anticoagulants such as apixaban are not currently available in Lesotho, but could represent a future option that would avoid the need for frequent monitoring (5). CONCLUSION The Motebang Hospital protocol for stroke care represents an evidence-based approach to reducing mortality, disability, and recurrence of a devastating and increasingly common disease. These practical guidelines designed for the Motebang Hospital can also serve as the basis for more general application to other district hospitals in Lesotho and other resource-limited settings. Table 1. Clinical features favouring ischaemic versus haemorrhagic stroke aetiology
Table 2 Emergency (Casualty) Care for Stroke Patients
Table 3. Inpatient Floor (Ward) Protocol for Stroke Patients
Table 4. Secondary stroke prevention
AUTISME ET ASPECTS ELECTROENCEPHALOGRAPHIQUES AU SERVICE DE NEUROLOGIE DU CENTRE HOSPITALIER NATIONAL UNIVERSITAIRE DE FANN DE DAKAR INTRODUCTION L’autisme se caractérise par trois symptômes majeurs : des altérations qualitatives des interactions sociales réciproques, des altérations qualitatives de la communication, des comportements, intérêts et activités réduits répétitifs et stéréotypés. Ces symptômes sont envahissants et atteignent tout le champ du développement (4,15). Au Sénégal, une étude sur l’infirmité motrice d’origine cérébrale avait retrouvé un syndrome autistique chez 7 sujets sur une série de 793 enfants (12). De nombreuses pathologies lui sont associées. Cinq à trente-huit pour cent des enfants autistes présentent une épilepsie comorbide (9,13,18) et selon certaines données (13) un pourcentage non négligeable d’enfants autistes développera des crises épileptiques à l’adolescence ou à l’âge adulte. Ainsi, des crises et syndromes épileptiques variables sont associés à l’autisme, et des anomalies épileptiques sont observées sur l’électroencéphalogramme (EEG) des sujets autistes même en l’absence de crise (3,9) pouvant suggérer un seuil épileptique bas (1). L’objectif de ce travail était de décrire les caractéristiques socio-démographiques des enfants autistes admis pour la réalisation d’un EEG, d’évaluer la fréquence des EEG anormaux chez les enfants atteints d’autisme sans épilepsie connue, et de décrire les caractéristiques des tracés EEG de ces enfants. Nous avons réalisé une étude rétrospective, transversale, et descriptive portant sur la période du 1er Janvier 2015 au 31 Octobre 2018, au laboratoire d’explorations fonctionnelles neurophysiologiques de la clinique de neurosciences Ibrahima Pierre Ndiaye du Centre Hospitalier National Universitaire de Fann. Nous avons passé en revue les enregistrements EEG qui ont été réalisés durant la période d’étude. Nous avons inclus dans l’étude tous les enregistrements EEG réalisés dont l’indication était l’autisme. Sur une fiche préétablie, nous avons relevé les données sociodémographiques ainsi que les caractéristiques morphologiques et topographiques (aspects normaux et pathologiques) du tracé EEG de veille et de sommeil. Les données ont été analysées grâce au logiciel Epi Info 7.2. Au total notre étude a porté sur 20 patients atteints d’autisme sur la période du 1er Janvier 2015 au 31 Octobre 2018. Il s’agissait de 13 enfants de genre masculin (65%) et 7 de genre féminin (35%) avec un sex-ratio de 2. L’âge des patients variait entre 3 et 10 ans, avec une moyenne de 4,5 ans ± 2,2. Un EEG de veille et de sommeil a été réalisé chez dix-huit patients (90%), tandis que 2 (10%) ont bénéficié uniquement d’un EEG de veille. Sur les enregistrements EEG de veille, le rythme de base était normal chez tous les patients, constitué d’un rythme alpha. Sur les enregistrements de sommeil, le rythme était constitué d’une activité thêta et delta bien organisée, et comportait les figures physiologiques du sommeil chez 16 patients (80%). La moitié des patients (50%) présentait des grapho-éléments pathologiques sur leur enregistrement EEG (tableau I). Ces derniers étaient constitués soit de pointes soit de pointes-ondes (tableau II). Ces grapho-éléments pathologiques avaient une distribution bilatérale asymétrique avec une prédominance droite chez 6 enfants (30%), bilatérale et symétrique chez 3 patients (15%) unilatérale chez 1 seul (5%). Les anomalies intéressaient les régions fronto-temporales chez 77,77% des sujets. La prévalence médiane mondiale de l’autisme est de 0,62 à 0,70% (6,7), bien que des estimations de 1 à 2% aient été faites lors des dernières enquêtes à grande échelle (11,16). Nous avons colligé 20 dossiers EEG de patients atteints d’autisme durant la période sur laquelle a porté notre étude, avec une prédominance masculine (65%) qui a été plusieurs fois rapportée dans la littérature (10,15,18). La prévalence des anomalies EEG infracliniques chez les enfants autistes, en l’absence de maladie épileptique connue, est rapportée à des taux variables dans les différents travaux de la littérature (4 à 86%) (10) avec des taux plus élevés que dans la population générale (10,13) Dans notre série, ce taux est de 50%, aussi bien dans le groupe des enfants ayant bénéficié d’un enregistrement de sommeil, que chez ceux qui ont bénéficié uniquement d’un enregistrement de veille. Des taux similaires ont été rapportés (10,18), mais également des valeurs beaucoup plus élevées telles que par Gubbay et al. (8), qui ont retrouvé un taux de 80% dans une série de 25 enfants atteints d’autisme, ainsi que par d’autres (3). La découverte de ces anomalies serait favorisée par l’enregistrement EEG en condition de sommeil (10,13). Les anomalies EEG décrites dans l’autisme ne semblent pas spécifiques (3,5) nous avons retrouvé des pointes et pointe-ondes, qui sont les grapho-éléments habituellement décrits (9,10). En dehors de ces anomalies de type épileptiques, d’autres anomalies non épileptiques ont été décrites (13) telles qu’une asymétrie de tracé ou un ralentissement. Nous n’avons pas retrouvé ces dernières, par contre une altération de l’architecture du sommeil a été mise en évidence chez 2 patients sur les 18 qui avaient bénéficié d’un EEG de sommeil. La distribution topographique des anomalies de l’EEG est variable chez les patients atteints de troubles du spectre autistiques (17,19). Dans notre étude, les signes irritatifs prédominaient dans les régions fronto-temporales (77,7% des patients), tout comme mentionné par la plupart des auteurs, pour qui les localisations prédominantes sont les régions périsylviennes (5,3,13,14). De même, la prédominance hémisphérique droite que nous avons relevé a également été mentionnée (13). En présence de ces anomalies, il est important de refaire un bilan clinique précis afin d’éliminer une épilepsie méconnue, du fait de la comorbidité reconnue entre épilepsie et troubles du spectre autistique (10). Ceci est d’autant plus important que certains déficits cognitifs seraient plus fréquents en cas de comorbidité, et qu’en outre l’épilepsie serait corrélée à des symptômes autistiques plus sévères (13). D’autre part, certains auteurs (5) pensent que les troubles comportementaux d’origine comitiale seraient largement sous-estimés dans les syndromes autistiques. Ainsi, une reconsidération des manifestations cliniques pourrait permettre de ne pas occulter des troubles comportementaux d’origine épileptique diagnostiqués à tort comme d’origine autistique. Par ailleurs, il est important de procéder à un suivi de ces patients car des études ont montré qu’un certain nombre parmi eux développent ultérieurement une épilepsie : 9.3% pour Parmeggiani A et al., 28.6% pour Kanemura H et al., cités par Precenzano et al. (13). En outre, il ne faudrait pas selon certains auteurs (2) se limiter aux explorations EEG standards qui peuvent occulter certaines anomalies à première vue, mais faire usage d’algorithmes qui sont plus aptes à refléter l’état de fonctionnement du cerveau et des connexions entre les différentes régions du cerveau. Concernant le traitement de ces anomalies, il n’y a pas de consensus (13). Selon des études, certains antiépileptiques voire les corticostéroïdes pourraient avoir un effet bénéfique sur ces anomalies EEG retrouvées en l’absence de maladie épileptique (13), sans qu’un argument consensuel définitif puisse être retenu. CONCLUSION Les relations entre autisme et épilepsie sont complexes mais leur compréhension pourrait permettre une meilleure connaissance des mécanismes physiopathologiques de l’autisme. Un nombre non négligeable de patients souffrant de troubles du spectre autistique, même sans épilepsie clinique, présentent sur leur enregistrement EEG des anomalies de type épileptique, avec une prédominance dans les régions périsylviennes. Il convient donc de réaliser un EEG précoce chez les patients autistes, du fait de la co-morbidité reconnue et des échanges de mauvais procédés rapportés. Pas de conflit d’intérêt
TABLEAUX Tableau I : Fréquence des grapho-éléments pathologiques sur l’électroencéphalogramme
Tableau II : Types de grapho-éléments pathologiques sur l’électroencéphalogramme
Articles récents
Commentaires récents
Archives
CatégoriesMéta |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
© 2002-2018 African Journal of Neurological Sciences.
All rights reserved. Terms of use.
Tous droits réservés. Termes d'Utilisation.
ISSN: 1992-2647