
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
RESUME La trypanosomiase humaine africaine (THA) ou maladie du sommeil, affection redoutable qui fit jadis beaucoup de ravages au sein des populations dans différentes régions d’Afrique dont celle de Nola en République Centrafricaine (RCA), reste un problème de santé publique en Afrique sub-saharienne. La cinquante sixième assemblée mondiale de la santé, tenue le 26/03/2003, reconnaissait que les douleurs, les souffrances et la mortalité dues à la trypanosomiase menacent quotidiennement plus de 60 millions d’habitants dans plus de 37 pays d’Afrique sub-saharienne dont 22 comptent parmi les moins avancés. Nous rapportons ici, l’histoire du foyer de THA de Nola en RCA, de 1971 à 2004. Sur le plan méthodologique, il s’agit d’une étude rétrospective couvrant la période de 1971 à 2004. Sur des fiches d’enquête établies, nous avons collecté des données à Nola à partir des registres des trypanosomés et à Bangui la capitale, à partir des rapports des missions de prospection de dépistage actif. L’analyse des données a été faite à l’aide du logiciel EPI INFO 6 version 2000. De 1971 à 2004, 3348 patients ont été recensés parmi lesquels 1814 anciens malades et 1534 nouveaux cas. Les femmes étaient plus atteintes (54%) que les hommes avec un sex-ratio de 1,2. La tranche d’âge la plus touchée est celle de 20 à 29 ans (67,0%). L’indice de morbidité nouvelle (IMN) est passé de 0,01% en 1971 à 1,7% en 1991 et à 0,05% en 2004. L’indice de contamination totale (ICT) est passé de 0,05% en 1971 à 2,3% en 1989 et à 0,05% en 2004. Les cas de rechutes et de réinfections représentaient 54,2% entre 1992 et 2004. La majorité des malades dépistés étaient en 2ème phase (64%). La lutte anti-vectorielle avec pose de pièges coniques imprégnés d’insecticides a été primordiale dans la maîtrise de l’épidémie dans cette région. Mots clés : Trypanosomose humaine africaine, histoire, république centrafricaine SUMMARY Human African Trypanosomiasis (HAT) or sleeping sickness, terrible affection which made major ravages in the past in different regions among which of Nola in Central African Republic (CAR), remains a public health problem in sub-saharan Africa. Key word: Human African trypanosomiasis, history, Central African Republic INTRODUCTION La trypanosomiase Humaine Africaine (THA) ou maladie du sommeil reste un problème de santé publique en Afrique sub-saharienne. Aujourd’hui l’Organisation Mondiale de la Santé (OMS) estime qu’entre 300 000 et 500 000 personnes sont touchées (16), mais seuls 10 à 15% de ces malades sont identifiés et traités (3). Cette maladie, jadis à l’origine de terribles épidémies, était presque éradiquée dans les années 1960. La République Centrafricaine (RCA) en 1945 avait le triste record de 25 000 trypanosomés sur les 50 000 que comptait l’Afrique Equatoriale Française (14, 15). La lutte menée par les secteurs des grandes endémies avait réduit ce nombre à 84 malades en 1968 pour l’ensemble des foyers (1). Mais elle a resurgi de manière dramatique en 1980 où on est passé de moins 80 malades annuels avant 1981 à près de 100 à 700 malades les années suivantes (1). L’objectif de notre travail est de décrire les données épidémiologiques de la THA du foyer de Nola de 1971 à 2004. Nous nous sommes attachés à décrire l’historique de cette maladie, à apprécier l’évolution de l’incidence et de la prévalence, à déterminer les raisons de la réémergence de cette pathologie à Nola et analyser les raisons de l’amélioration récente de la situation épidémiologique dans cette région du pays. METHODOLOGIE Il s’agit d’une étude rétrospective couvrant la période de 1971 à 2004. Sur des fiches d’enquête établies, nous avons collecté les données à partir des registres des trypanosomés de l’hôpital préfectoral de Nola, et à partir des rapports de missions de prospection de dépistage actif disponibles au bureau du Programme National de Lutte contre la Trypanosomiase Humaine Africaine (PNLTHA). Les variables collectées pendant l’étude sont en rapport avec l’identification du malade (âge, sexe, localité, année), le dépistage (dépistage actif ou passif, ancien ou nouveau cas), la biologie du malade (CATT, recherche de trypanosome), le traitement (pentamidine, mélarsoprol), l’évolution (guérison, rechute, décès) et les prospections (effectifs des malades et de la population visitée). RESULTATS La RCA comptait à l’époque quatre foyers de THA dont le plus important était le foyer de Nola (figure 1). DISCUSSION La RCA a connu une longue période d’accalmie au lendemain de l’indépendance (1960-1978). Pendant cette période on a assisté à une défaillance de la surveillance épidémiologique marquée par des prospections et des dépistages partiels. Ce qui aura pour conséquence le réveil en 1979 du foyer de Nola qui était presque éteint (17). La flambée épidémiologique survenue entre les années 1980 et 2000 s’observait dans d’autres foyers de la RCA (14,15), mais aussi dans d’autres régions d’Afrique centrale telles qu’au Congo (20) et en République Démocratique du Congo (4,13). Cette flambée s’explique par la détérioration des systèmes de santé, l’abandon ou l’affaiblissement des programmes de lutte, l’instabilité sociopolitique avec les mouvements incessants de la population. Tous ces éléments ont entraîné la création en 1990 du Programme National de Lutte contre la Trypanosomiase Humaine Africaine (PNLTHA) par le Ministère de la santé publique et de la population (MSPP) de la RCA. Ce programme a uvré efficacement à la stabilisation puis à la décroissance progressive de la maladie, par l’intensification de la prospection (dépistage actif et traitement), la création des centres de traitement ambulatoire (Nola centre, commune de Bilolo) la lutte anti-vectorielle avec pose de pièges coniques imprégnés d’insecticides (18). Ceci nous permet de dire que si aucune mesure n’est prise contre les glossines, il persistera des personnes infectées pouvant transmettre les trypanosomes quelle que soit la qualité des prospections. Le foyer de Nola comporte trois zones d’infestation dont l’épicentre de la maladie se trouve dans la commune de Bilolo qui à celle seule a déclaré 59% du total des cas. Ceci pourrait s’expliquer par les conditions favorables liés à l’environnement naturel (10). Les sujets âgés de moins de 30 ans représentaient la tranche la plus affectée avec 67% des cas. Ce résultat corrobore ceux rapportés en Ouganda (11) et à Kinshasa (4). Cette situation peut s’expliquer par le fait qu’une grande majorité de la population était composée de jeunes qui travaillent dans les mines d’or et de diamant, dans les champs et qui font la pêche. Ce qui augmente leur exposition aux piqûres des glossines. Nous avons retrouvé une forte proportion de trypanosomiase infantile (18,1%) qui serait due probablement au fait que ces enfants accompagnent leurs parents dans leurs besognes. La prédominance féminine (54,2%) retrouvée avec un sex-ratio de 1,2 a été décrite par d’autres auteurs (5, 7, 8). Elle s’expliquerait par le fait que ce groupe de population s’occupe plus des activités ménagères, champêtres et de pêche (par petit barrage). Ce qui augmente le temps de contact être humain-mouche tsé-tsé. A l’inverse, la plus faible atteinte des hommes pourrait s’expliquer par le phénomène d’immigration temporaire pour des raisons d’emploi ou de scolarité, comme l’évoquait FOURNIER et al. (6), ce qui diminue le risque d’exposition à la THA. Plusieurs raisons peuvent expliquer le réémergence de la THA à Nola. On peut évoquer soit l’existence d’un environnement naturel permettant la persistance et l’éclosion de la maladie dans ce foyer. Ce phénomène a été évoqué au Soudan (3). Soit la modification de l’écosystème pour l’implantation des sociétés forestières à partir de 1969, entraînant le redéploiement des mouches autour des habitats. Ceci corrobore les travaux de LAVEISSIERE et al. (12) en Côte d’Ivoire. Soit l’existence d’un réservoir animal déjà évoqué par SCHNEIDER en 1944 lorsqu’il avait découvert des trypanosomes dans le sang de certains animaux (15) dans le village de Bilolo. Mais cette probabilité de transmission de l’animal à l’homme serait mineure, néanmoins elle permettrait à la maladie de ne jamais tout à fait disparaître. On peut ajouter la défaillance de la méthodologie de lutte qui ne permet pas d’attaquer dans son ensemble la chaîne épidémiologique de la maladie, notamment les dépistages partiels et inefficaces, et l’absence de la lutte antivectorielle. CONCLUSION L’évolution de la THA dans le foyer de Nola de 1971 à 2004 est caractérisée par une période d’accalmie, une période de flambée épidémiologique et une période d’amélioration de la situation épidémiologique après la création du PNLTHA en 1990. Dès lors, l’intensification de la stratégie de la lutte, l’implication de la communauté dans l’action de lutte anti-vectorielle, et la contribution des institutions internationales ont été déterminantes dans l’amélioration récente de la situation épidémiologique. Seul le maintien d’une surveillance efficace de toute la chaîne épidémiologique permettra de conserver les acquis de toutes ces années de lutte.  Figure 1  Figure 2  Figure 3  Figure 4
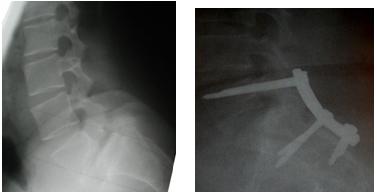
RESUME Objectif Méthode Résultats Conclusion Mots clés : Afrique, lyse isthmique, Niger, spondylolisthésis. Summary Objective Results Conclusion Key words: Isthmic spondylolisthesis ,Niger, spondylolisthesis. INTRODUCTION Le spondylolisthésis correspond à un glissement en avant d’un corps vertébral accompagné de ses pédicules, ses apophyses transverses et ses articulaires supérieures. Ce glissement est rendu possible par une solution de continuité ou une élongation de la portion interarticulaire de l’arc postérieur ou isthme. (6). Néanmoins certaines études ont rapportées des cas de spondylolisthésis sans lyse ni élongation isthmique (4,13). Taillard (23) avait classé les spondylolisthésis en 4 théories : congénitale, traumatique, trophostatique et dysplasique.Une autre classification basée sur les travaux de Newman et Wiltse (14, 25, 26, 27) classent les spondylolisthésis en 5 types : isthmique, dégénératif, dysplasique, traumatique et pathologique. Le spondylolisthésis dégénératif est une pathologie de l’adulte au dessus de 40ans (19,21, 22, 4). Le spondylolisthésis par lyse isthmique est une pathologie de l’adolescent (20,25). La solution de continuité peut être à l’origine d’un glissement. Ce glissement est une recherche d’équilibre selon certains auteurs (21,24). Les signes cliniques du spondylolisthésis par lyse isthmique sont faits de lombalgies isolées ou associées à des radiculalgies. Le traitement initial est conservateur surtout si les signes cliniques prédominants sont les lombalgies. La présence des signes radiculaires ou de lombalgies invalidantes et rebelles constituent une indication du traitement chirurgical (11, 16). METHODE Il s’agissait d’une étude prospective en série continue réalisée dans le service de neurochirurgie de l’hôpital national de Niamey entre Janvier 1999 et Janvier 2004. Tableau 1 : cotation de Beaujon. Ainsi pour chaque patient on attribuait en préopératoire, une note sur 20 qui est d’autant plus basse que l’état fonctionnel est mauvais. Cette note est obtenue en faisant la somme des 7 paramètres dont la valeur maximale est de 2, 3 ou 4 suivant le paramètre. Le total maximal possible de 20 points correspond à l’état fonctionnel parfait. Ainsi le gain post opératoire est la différence entre le score préopératoire et le score post opératoire et le gain relatif correspond au rapport du gain postopératoire sur le gain maximum qui était théoriquement possible. Pour l’évaluation radiologique les patients étaient classés selon la classification de Meyerdig (10). RESULTATS La série était constituée de 11 hommes et 09 femmes. La moyenne d’âge était de 27ans avec des extrêmes de 17ans et de 40 ans. Sur le plan professionnel il s’agissait de 7 ouvriers agricoles, 2 élèves, 3 employés de bureau, 1médecin, 1avocat, 2 infirmières, 2 enseignants, 2 manutentionnaires. Les lombalgies évoluaient depuis 3,5 ans en moyenne et les radiculalgies depuis 1,01 an en moyenne. Tous les patients admis dans cette étude souffraient de lombalgies associées à des radiculalgies à l’exception d’une jeune élève de 17ans avec des lombalgies invalidantes et rebelles. Ces radiculalgies étaient L4 et L5 bilatérales pour 1patient sur 20 ; L5 et S1 bilatérales pour 1/20 patients ; L5 unilatérale chez 5/20 ; L5 bilatérales chez 4/20 ; S1 unilatérales chez 1/20 ; S1 bilatérales chez 1/20 ; elles étaient mal systématisées bilatérales chez 7/20 patients. Sur le plan radiologique le spondilolysthésis était au stade I pour 10 patients, au stade II pour 7 patients et au stade III pour 3 patients. La lyse isthmique était sur L5 bilatérale chez 17cas patients (85% des cas) ; sur L4 dans 3 cas (15%). Le score moyen préopératoire est de 9.1 sur 20. Les résultats de la cotation de Beaujon à 3 mois en postopératoire étaient les suivants : DISCUSSION Le spondylolisthésis par lyse isthmique est une pathologie de l’espèce humaine en relation avec le développement de la lordose lombaire. Le cas le plus précoce a été rapporté chez un nourrisson de 3.5 mois (1). Pour la plupart des auteurs c’est une pathologie de l’enfant et l’adolescent. (17,19, 21, 25). La moyenne d’âge varie entre 25ans et 30ans. Dans cette série la moyenne d’âge est de 27 ans avec des extrêmes de 17 ans et de 40ans. Les anomalies dysplasiques lombaires et sacrées associées au développement de la lordose lombaire lors de la mise en position débout entraînent dans certains cas une recherche d’équilibre qui conduit à la lyse isthmique et éventuellement au spondylolisthésis. Cette hypothèse expliquerait la précocité de la survenue de la lyse isthmique. (7, 9, 25). Dans la littérature on ne retrouve pas de relation entre le sexe et le spondylolisthésis; dans cette série, il y a un équilibre dans la répartition entre les deux sexes (11 hommes et 9 femmes). Dans la population européenne il y a 5 à 7% des sujets présentant une lyse isthmique (19, 21). Cette proportion serait légèrement supérieure chez les japonais, les bantous et les esquimaux (21). Sur le plan professionnel plusieurs auteurs ont rapporté une relation avec une activité sportive intense et la découverte d’une lyse isthmique avec ou sans glissement (9,19, 21).Certains auteurs par contre rapportent que l’activité sportive chez un adolescent ayant une lyse isthmique n’est pas forcement un facteur aggravant (12). Dans cette série 45% des patients exerçaient une activité physique intense du fait de leur profession et un patient était un grand sportif. Etaient inclus dans cette série que les patients avec des lomboradiculalgies invalidantes rebelles ou devenues déficitaires à l’exception d’un cas. L’évolution de la lyse isthmique vers les spondylolisthésis ne se fait que dans 50 à 65 % des cas (1, 21). La radiculalgie est un signe qui apparaît au cours de l’évolution de la lyse isthmique du fait de la formation des nodules de Gill (3). Cette radiculalgie est plus fréquente en cas de spondylolisthésis. Dans cette série ces radiculalgies sont présentes chez 95% des patients. Ceci pourrait s’expliquer par l’absence de traitement conservateur précoce du fait du retard à la consultation. La racine la plus atteinte est L5 (9/20) et en cas de déficit elle est aussi la plus touchée (5/9) ; elle est suivie par la racine S1 (8/20) puis L4 (3/20). Aucun trouble sphinctérien n’a été rapporté dans cette série. La prédominance de la lyse isthmique sur L5 est rapportée dans la littérature (16,19, 21, 25). Selon Ganju (1) la lyse isthmique est sur L5 dans 90% des cas, sur L4 dans 5% des cas et ailleurs dans 5% des cas. Dans la littérature le traitement chirurgical concerne une minorité de patients après échec du traitement conservateur. Pour la majorité des auteurs (1, 2, 4), 4 critères sont admis pour l’indication opératoire dans le spondylolisthésis par lyse isthmique : persistance de la douleur et des signes neurologiques malgré un traitement conservateur bien conduit (2), progression du glissement supérieur à 30% (5), grade Meyerdig supérieur à II (4), déformation lombosacrée secondaire avec troubles de la marche (4). Dans notre série la douleur radiculaire rebelle, invalidante ou déficitaire a été l’élément déterminant dans l’indication opératoire. En présence de signes de sténose lombaire le traitement chirurgical est meilleur sur la régression de la douleur et la récupération fonctionnelle que le traitement conservateur (6). Selon les séries le traitement chirurgical peut être une décompression, une greffe in situ, une instrumentation avec greffe ou une chirurgie de réduction. La décompression seule par l’intervention de Gill (3) peur être responsable d’un glissement secondaire dans 23% des cas selon Osterman (15). Cette technique est rarement proposé à l’enfant car source d’instabilité (1). Pour les lombalgies de l’adulte, surtout avec progression du glissement ou les lomboradiculalgies du jeune rebelles et invalidantes, des techniques de greffe sont indiquées : greffe intersomatique antérieure, greffe inter somatique postérieure, transforaminale, inter transverse etc. Les résultats sont satisfaisants sur la douleur (18). Dans les spondylolisthésis de grade I un abord direct de la lyse isthmique peut être réalisé par cerclage intertransversaire ou par vissage isthmo-pédiculaire. L’avantage de ces abords directs est la préservation des segments adjacents (1). Dans les spondylolisthésis de grade élevé le vissage pédiculaire avec instrumentation offre une meilleure stabilité mécanique que les autres techniques de fixation (1). Les techniques de réduction avec pour objectif d’obtenir une correction des déformations, d’obtenir un réalignement pour une meilleure fusion comporte des risques neurologique pouvant atteindre 20% des cas (1). Une bonne fusion peut être obtenue par un abord antérieur avec greffe inter somatique. Un montage semi-rigide court associé ou non à une chirurgie discale par voie antérieure ou postérieure avec greffe ou cage inter-somatique est recommandé par plusieurs auteurs, surtout lorsque la hauteur du disque est conservée (8, 9,12). Notre attitude thérapeutique dans cette série a consisté en une décompression radiculaire puis vissage pédiculaire sur montage rigide court car l’objectif principal était la sédation de la radiculalgie. Les résultats post opératoires sont bons ou très sur le plan fonctionnel bons pour 15 patients sur 20. Le gain postopératoire varie de 2 à 13 avec un gain moyen de 7. Le gain relatif moyen est de 45%. Les résultats chirurgicaux des formes symptomatiques avec des signes radiculaires sont bons ou très bons dans plusieurs études (6, 25).Une étude multicentrique randomisée (6) a évaluée les résultats du traitement chirurgical (laminectomie avec ou sans fusion) dans les spondylolisthésis avec sténose lombaire. Le medical out come study (36 items avec 100 points scales) et l’Oswestry disability index (100 points scales) ont été utilisés comme table d’évaluation. Cette étude a montré une amélioration significative de la douleur et une récupération fonctionnelle significative. CONCLUSION Le spondylolisthésis par lyse isthmique est une pathologie de l’adolescent et de l’adulte jeune à Niamey. La recherche de la lyse isthmique dans le bilan de la lombalgie de l’adulte jeune et de l’adolescent doit être systématique. Le traitement conservateur est suffisant lorsque les lombalgies ne sont pas invalidantes et en l’absence de signes radiculaires. Tableau 1 : cotation de Beaujon
 Image 1
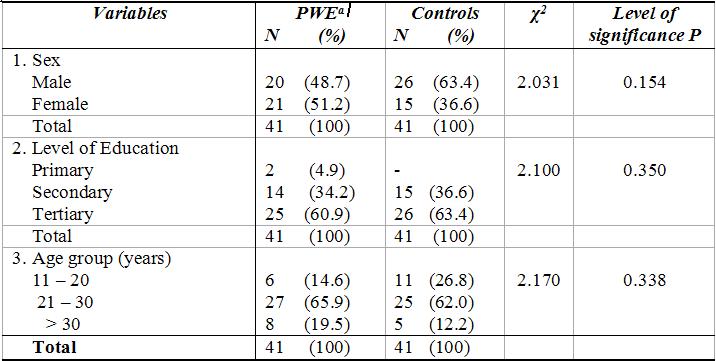
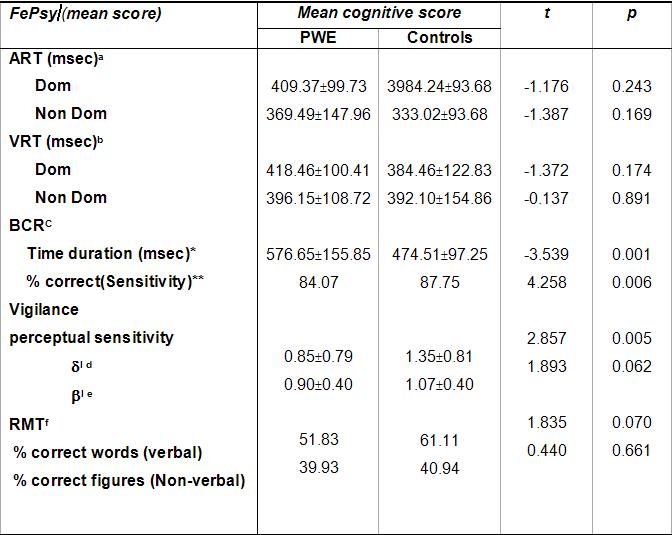
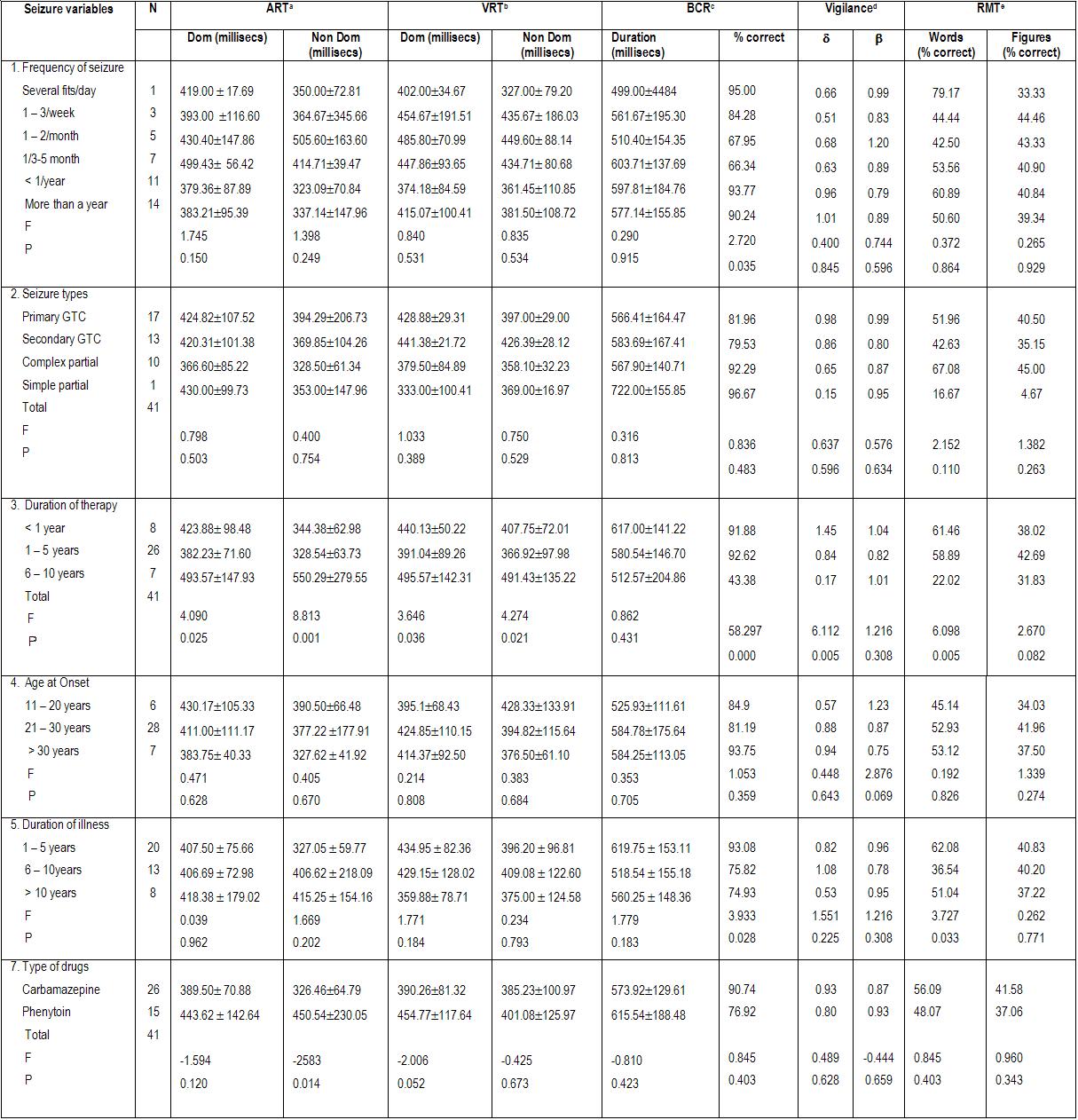
ABSTRACT Background and Purpose Methods Results Key words: cognitive, epilepsy, memory, mental speed, Nigeria. INTRODUCTION Epilepsy is the second most common disorder of the central nervous system, affecting 1% of the human population (3). It is particularly highly prevalent in developing African countries (24,28). The concern over the quality of life of patients with epilepsy (PWE) has attracted many attempts at elucidating the impact of seizure variables like seizure types, frequency of seizures, duration of epilepsy, and effect of anti-epileptic drugs (AEDs) on the cognitive functioning of these patients. Several reports have confirmed the presence of cognitive disturbances in PWE and the consequences on their day-to-day activities (1, 9, 20, 29, 30). Cognitive deterioration observed in epilepsy may arise ab initio as a result of the epilepsy as the same brain dysfunction that causes the seizures can also lead to learning disabilities, memory impairment and psychomotor retardation (1,24,28) or it may be secondary to anti-epileptic medications which can exacerbate these cognitive disturbances (9,29). Several reports have shown that cognitive deterioration is worse among patients with generalized seizures than among those with partial seizures (12,15). This explains the contribution of cognitive impairment to poor quality of life among these patients with generalized epilepsies. The longer the duration of epilepsy the higher the likelihood of cognitive deterioration (6, 14) though it has been reported that duration of epilepsy has no effect on cognition (13). Though cognitive disturbances have been noted in newly diagnosed PWE (20, 26), but more importantly is the contribution of the anti-epileptic drugs (AEDs). Anti-epileptic drugs can also contribute to cognitive impairments especially those drugs with significant sedative effects. Phenobarbitone, diazepam, phenytoin, sodium valproate and carbamazepine have all been shown to affect the cognitive abilities of PWE but in differing degrees (9, 29, 31). The effect on cognition is worse with phenobarbitone and diazepam (25, 29). Anti-epileptic drugs (AEDs) may cause impairments in cognitive functions such as attention, memory and psychomotor speed. There is evidence that drug-induced cognitive impairment has great impact on critical daily life function of patients with epilepsy (1,9). The cognitive effects of AEDs are of special concern because they are iatrogenically induced (1). Comparative studies of AEDs in developed countries have shown that while carbamazepine had minimal side effects, phenytoin and phenobarbitone have less favorable cognitive side effects with impairments of mental speed, memory and attention (29, 30,31). These latter two drugs are however the most widely used in developing countries, because of good efficacy, broad spectrum of activity and low cost (10,19). This study was designed to assess the effects of seizure variables, namely; duration of epilepsy, frequency of seizures, seizure types, type of anti-epileptic medication, duration of AED use and age at onset of seizures on the cognitive performances of our patients with epilepsy. A computerized cognitive assessment test battery, the FePsy(16) which has been validated earlier among Nigerians (21) was used to assess the memory, attention, concentration and psychomotor speed of the patients. PATIENTS AND METHODS The cohort comprised of forty-one patients with epilepsy (PWE) and forty-one sex-, age-, level of education- and socio-economic-matched controls. The study participants were adult patients attending the Neurology clinic of Obafemi Awolowo University Teaching Hospitals Complex, Ile-Ife, a tertiary health facility situated in metropolitan Nigeria. Informed consent was obtained from each subject before commencement of the study. The ethical committee of the hospital gave approval for the study. The inclusion criteria included patients aged between 16 and 55years with history of epilepsy as defined by the characteristic eye witness of at least two separate unprovoked non-febrile seizures. Patient must have also completed at least primary school education (some of the test items require subjects to be literate) and must be on phenytoin or carbamazepine therapy only to avoid the confounding effects of polytherapy. These two drugs are the most commonly used anti-epileptic drugs in our center. The exclusion criteria included use of antiepileptic drugs other than phenytoin or carbamazepine, seizures secondary to other causes (such as fever, infection, head injury, cerebrovascular disease), patients with visual and/or hearing impairment, musculoskeletal abnormalities and seizures occurring 24 hours preceding cognitive functions assessment. The controls were age, sex, education and social status matched with PWE and they had no family history of seizures. A questionnaire was developed and applied to all subjects. History was taken from patients and eye witnesses. Emphasis was placed on demographic data (age, sex, level of education) and details of seizure variables which include duration of seizures, seizure types, type of AED administered and duration of drug therapy, age at onset of epilepsy and frequency of seizures. The history of birth, perinatal injury, fever, head injury and drug abuse was sought. Detailed general physical examination was carried out on each subject and the seizure types were classified according to the International League against Epilepsy (ILAE 1981) criteria (21). The cognitive performances of the PWE and controls were assessed with the computerized cognitive assessment test battery, the FePsy (19). Electroencephalographic (EEG) studies were done in all the patients. The differences in sex, age and level of education of the PWE and controls were tested for statistical significance with the chi-square test. The difference in the cognitive performances of the PWE and controls was analyzed using Student t test. The impact of seizure variables on cognitive performances in PWE was analyzed using 2-way Analysis of Variance (ANOVA). The level of significance was taken as p <0.05. RESULTS The study participants comprised 41 PWE and 41 controls with mean ages of 28.32(± 9.22) years and 25.95(± 7.72) years respectively. There were 20 males and 21 females in the PWE group and 26 males and 15 females in the control group. Other demographic details of the study participants are as outlined in table 1. Majority of the PWE had generalized epilepsies (73.2%) with approximately half of this group having secondarily generalized seizures. Complex partial seizures were found to be commoner than simple partial seizures. Most of the patients (65.9%) presented in the second and third decades of life. More than half (51.2%) of the patients who participated in the study had had seizures for more than five years. Sixteen of the patients (39%) had frequent seizures i.e. seizures in excess of more than or equal to one fit in 3-6 months. Carbamazepine was used in most of the patients to achieve seizure control (66.7%). The cognitive performances of the patients with epilepsy (PWE) and the controls are presented in table 3. The PWE had prolonged response time in both auditory and visual reaction tasks when compared to the controls but this difference did not reach statistical significance (p>0.05). There was however a significant difference in the psychomotor speed of the PWE and that of the controls using the complex reaction task (binary choice reaction task). In addition, the concentration ability of the PWE was significantly impaired as shown by their lower percentage of correct responses (p<0.01). The PWE were significantly impaired in their attention span (p=0.005) but not in their recall abilities (p= 0.07). The effects of the seizure variables on the various cognitive tasks performance are presented on table 4. The frequency of seizures and the type of epilepsy did not affect the cognitive performances of the PWE significantly with the exception of their concentration abilities which was negatively impacted by the seizure frequency using the binary choice reaction task (p=0.035). The duration of epilepsy significantly affected the concentration abilities and the verbal memory performance of the PWE negatively (p<0.05) but had no statistically significant effect on other cognitive tasks. The age at onset of epilepsy did not show any appreciable significant effect on their cognitive functioning. The type of medication i.e. AED administered to the patient had no effect on the cognitive performance with the exception of the auditory reaction time which was significantly prolonged using the dominant hand (p=0.014) among PWE on phenytoin. The duration of therapy however significantly affected their reaction time (both auditory and visual), concentration and attention, and their information recall abilities (p<0.05). DISCUSSION Cognitive deterioration in epilepsy has been associated with poor quality of life, marital disharmony and family breakup, inability to secure employment, retrenchment from work, academic disruption and social stigma (5). Most patients with epilepsy report memory impairment during clinic visits as this obviously disrupt their academic performance (20,27). Cognitive dysfunctions which have been associated with epilepsy include memory impairment, psychomotor retardation, inattention and lack of concentration, reduced motor speed and in patients with significant brain damage impaired visual scanning task (1,9,12,20,26). In this study the lack of significant difference in psychomotor and recognition memory performances of the PWE and controls using the auditory and visual simple reaction time tasks is inconsistent with previously documented observations in the literature. This may be as a result of the type of cognitive tests used or the small sample size, though we opined that it is likely due to the latter. A previous publication by Ogunrin et al (20) that studied a larger sample size and used similar cognitive tests showed significant difference between PWE and controls. Despite the fact that the etiology of the epilepsy may cause cognitive disruption by itself, the contributory roles of various seizure variables need evaluation. This study showed inconsistent pattern when effects of seizure frequency and epilepsy type on cognitive performance were assessed. The seizure frequency affected concentration ability but had no effect on mental speed, an observation that is not consistent with most reports in the literature. The lack of effect was noted when the simple reaction tasks were used to assess the (psychomotor) mental speed of the patients. Furthermore, the pattern of performance showed a faster response with the non-dominant hands for the auditory (simple) reaction task, an observation which has been reported earlier and is different with what was observed by researchers in the western countries (20). The negative impact on mental speed was however noted with complex (binary) reaction task. This suggests that more complex cognitive tasks may be more demanding and difficult to cope with in epilepsy. The same pattern was observed for duration of epilepsy and type of medication used. It has been established that the higher the frequency of seizures the more severe the cognitive dysfunctions thus underscoring the importance of effective and optimal seizure control with appropriate AEDs (2,27). Evidences from the literature clearly show that the more frequent the attacks of seizures, the more the likelihood of impaired cognition for both idiopathic and symptomatic epilepsies (1,30). It is likely that recurrent seizures with loss of consciousness directly results in cognitive disturbances by interfering with learning, and indirectly by causing cerebral damage, especially in patients with chronic epilepsy (1,7,12,14). A direct relationship between the number of seizure attacks and cell loss in the hippocampus, an essential structure in some memory processes, has been reported (17). Twin studies have also shown that the twin with more frequent seizures performed worse on tests of cognitive functions (7). The degree of seizure control thus appears to be important as good control has been associated with better performance (18). The age at onset of seizures and the seizure type appeared to have no significant effects on the cognitive abilities of the PWE. This is at variance with earlier observations (6, 14, 15,18). Early onset of seizures is recognized to have deleterious effect on cognition (27). Since childhood is an important period during which one starts learning, then onset of epilepsy during childhood interferes greatly with the acquisition of learning abilities. Many authors have noted that the onset of seizures in early childhood is associated with a higher risk of cognitive impairment than when seizures started in late childhood or the teenage years. Attempts to associate seizure types with cognitive function have not yielded entirely consistent results. Generalised absence seizures have been reported to be less damaging than generalised tonic- clonic seizures, and patients with partial seizures performed better on tests of cognitive tasks than those with generalised seizures (6,27). The cognitive performance was however worse in PWE on phenytoin when compared with those on carbamazepine. This pattern has been consistently noted by other authors (8,23,29,31). The duration of therapy affected all the cognitive tasks assessed portraying the contributory role of prolonged administration of AED on cognitive functioning of PWE. It is important to emphasize that the inconsistencies observed with this study may not be unrelated to the small sample size. Most of the earlier studies among Nigerian Africans assessed larger sample sizes of PWE. Apart from the limitation of small sample size, the other limitation of this study is our inability to assess the serum levels of the AEDs (carbamazepine and phenytoin) in these patients.
 Table 2: Seizure variables in PWE
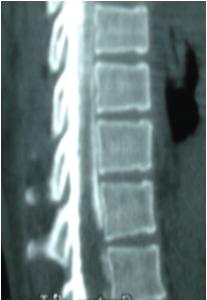
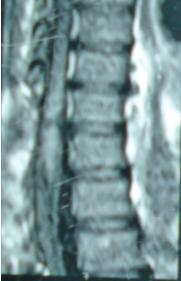
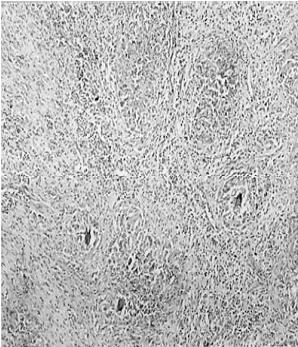
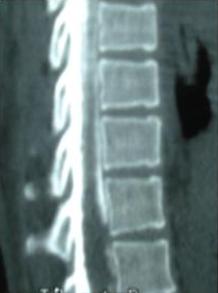
1 – MDS 13th International Congress of Parkinson’s Disease and Movement Disorders June 7-11, 2009 Paris, France For more information: www.movementdisorders.org/congress/congress09 – 2 – WFNS Meetings. Boston Congress Aug. 30 – Sept. 4, 2009 For more information: http://www.wfns.org/pages/wfns_boston_2009_world_congress_brochure/110.php – LE TUBERCULOME INTRA MEDULLAIRE : UNE CAUSE RARE DE PARAPARESIERESUME Le tuberculome intra-médullaire (TIM) est une localisation de la tuberculose du système nerveux central. Nous rapportons un cas de TIM, chez un patient de 48 ans, sans antécédents particuliers, qui a consulté pour un déficit moteur des 2 membres inférieurs d’installation progressive. L’examen clinque a permis d’objectiver un syndrome de compression médullaire thoracique. La découverte d’une masse intra médullaire, après les explorations neuroradiologiques (myéloscanner et IRM), nous a fait poser l’indication d’une exérèse micro-chirurgicale. Le diagnostique de TIM a été affirmé par l’examen anatomo-pathologique de la pièce opératoire. L’association d’une chimiothérapie anti-tuberculeuse au delà de 6 mois après l’exérèse chirurgicale, à permis une guérison complète après un recul de 18 mois. Mots clés : tuberculome, lésion intra médullaire, chirurgie ABSTRACT The intra-medullary tuberculoma (IMT) is a rare tuberculosis located in the central nervous system. We report a case of IMT in a 48-year-old man with a progressive paresis of the lower limbs. The patient had previously been healthy. The neuroradiologic investigations showed an intra-medullary mass. A total exeresis was performed via a micro-surgical approach. The IMT diagnosis was confirmed by the anatomo-pathological examination.The association of an anti-tuberculosis chemotherapy for 6 months after the surgery led to a complete healing after an 18 months lapse. keys words : intramedullary lesion – spinal cord – tuberculoma INTRODUCTION La tuberculose constitue un véritable problème de santé publique dans le monde, mais surtout dans les pays d’Afrique et d’Asie [2, 5, 7, 8, 10, 11, 15, 17,18]. En outre, la pandémie du SIDA, a favorisé au cours de ces dernières décennies, une recrudescence des cas de tuberculose. La localisation au niveau du système nerveux (SNC) est rare ; Elle représente environ 0,5 à 2 % des patients tuberculeux [5, 8, 14, 15,16]. Le tuberculome du SNC est encore plus rare, il se développe le plus souvent dans le parenchyme cérébral. La location intra-médullaire du tuberculome représente 0.02% des atteintes tuberculeuses du SNC [1,2,5,6,8,9,14,15,16 ]. Le tuberculome intra-médullaire (TIM) a été très peu rapporté dans Littérature [9]. Il se manifeste habituellement par un tableau de compression médullaire lente, et les progrès L’IRM permettent aujourd’hui de mieux préciser ses caractéristiques radiologiques. Nous rapportons le seul cas observé dans le service de Neurochirurgie d’Abidjan. Le diagnostic de TIM a été fait par l’examen anatomo-pathologique de la pièce opératoire après la chirurgie. Les aspects épidémiologiques, cliniques, radiologiques et les modalités thérapeutiques sont discutés à l’aide de la littérature. OBSERVATION Monsieur O.A, âgé de 48 ans, à consulté pour des troubles de la marche à type de fatigabilité, d’évolution aggravante depuis 2 mois. Il se plaignait des paresthésies, de dorsalgies et de troubles vésico-sphinctériens d’apparition récents à type de dysurie. La symptomatologie évoluait dans un contexte de subfébrile, avec un amaigrissement progressif. Ses antécédents médicaux étaient sans particularités. Il n’y avait pas de notion de contage tuberculeux. L’examen clinique retrouvait un état général relativement bien conservé, Une para parésie spastique cotée à 4/5 droite et 3/5 à gauche, une hypo-esthésie à niveau T12. Les reflexes rotuliens et achilléens étaient vifs et il existait un signe de Babinski bilatéral. Le bilan biologique montrait une anémie normochrome normocytaire, une vitesse de sédimentation légèrement accélérée, Le bilan rénal était sans particularité, et la sérologie HIV était négative. Un myéloscanner thoracique initialement réalisé montrait un élargissement du cône médullaire évoquant la présence d’une tumeur intramédullaire (figure 1). L’IRM visualisait la lésion sous forme de signal hétérogène avec une composante périphérique se rehaussant après injection du produit de contraste et centrée par une hypo-intense (figure 2). On retrouvait également un signal hypo intense sur les différentes séquences, à la périphérie de la lésion, témoignant de l’existence d’un dème péri-lésionnel. Le diagnostique d’astrocytome intra médullaire était évoqué et l’indication de l’exérèse microchirurgicale était retenu. Une corticothérapie a été instituée avant la chirurgie. Une laminectomie de T12-T11 a été réalisée. Il n’y avait aucune anomalie extradurale en dehors d’une expansion du cône médullaire témoignant de la présence d’une masse intradurale. Sous microscope opératoire, une myélotomie longitudinale permettait l’exérèse en monobloc, d’une masse ovalaire mesurant 3X2X1 cm, d’aspect jaunâtre, de consistance ferme et granuleuse. Cette lésion se décollait facilement du tissu nerveux. L’examen anatomopathologique de la pièce opératoire mettait en évidence une lésion folliculaire centrée par une nécrose caséeuse plus ou moins complète entourée de cellules épithélioïdes, de cellules géantes de Langhans et des lymphocytes en périphérie. La coloration de Ziehl-Nielsen n’a pas permis d’isoler des Bacilles Acido-Alcoolo-Résistants (BAAR). Le diagnostic de TIM a alors été retenu (Figure 3). L’évolution post-opératoire a permis de constater à J3 une récupération partielle du déficit moteur et des troubles vésico-sphinctériens. Le patient a été mis sous traitement anti-tuberculeux pendant 10 mois et des séances de rééducation fonctionnelle ont été prescrites. L’évolution à 18 mois était satisfaisante avec une reprise autonome de la marche .L’IRM de contrôle n’a montré pas de récidive de la lésion tuberculeuse. DISCUSSION L’atteinte médullaire de la tuberculose est le plus souvent secondairement à une localisation vertébrale ; c’est la classique spondylodiscite à BK ou mal de POTT. Par ailleurs, le Mycobactérium tuberculeux peut atteindre directement le tissu nerveux. Les lésions tuberculeuses vont se développer soit dans l’espace épidural ou dans l’espace sous-dural par diffusion à travers les méninges, soit dans le parenchyme cérébral ou médullaire. Il se produit une réaction inflammatoire locale qui va évoluer vers la formation du granulome. [ 7,13]. La localisation du tuberculome dans le parenchyme cérébral est plus fréquente que dans le tissu médullaire, avec un ratio de 1/42 [ 2,3,4,8,15,17,18 ]. Le rapport de poids, et la différence de vascularisation, plus importante au niveau cérébral, pourraient expliquer cette disproportion [3, 9,17]. Le tableau clinique de la TIM est semblable à celui d’autres tumeurs intra-médullaires [17]. Il s’agit généralement d’un syndrome de compression médullaire lente dont l’expression clinique est fonction du siège de lésion [5,7,9,15,16]. La survenue d’un déficit moteur d’aggravation rapide a été rapportée par certains auteurs [9,18]. L’association de signes d’imprégnations tuberculeuses tels que la fièvre, les sueurs nocturnes, et la notion de contage tubage tuberculeux peuvent être retrouvés à l’interrogatoire [5]. Dans notre cas, il s agissait d’un déficit moteur d’installation lente dans un contexte d’amaigrissement et de fièvre vespérale. La myélographie et le myéloscanner permettent de mettre en évidence une image de grosse moelle avec la présence d’une lésion intra-médullaire [6,15,18], mais ces examens sont aujourd’hui de moins en moins réalisés. L’IRM précise mieux les caractéristiques radiologiques de la TIM, permettant une approche diagnostique plus aisée [2,3,4,5,7,8,9,14,16]. Rhoton [12] a été le premier à décrire ces caractéristiques en 1988. actuellement, avec les progrès de l’IRM, deux aspects du TIM sont décrits en fonction du stade évolutif [3,5,6,15,16,17] . Au stade initial, il existe une réaction inflammatoire avec un dème périphérique plus ou important, la capsule est pauvre en collagène , et le tuberculome apparaît iso-intense aussi bien en séquence T 1 et T2 avec un rehaussement homogène avec après injection. A un stade plus tardif, la capsule du tuberculome s’enrichit en collagène et la réaction inflammatoire circulaire décroit en intensité ou disparaît. La lésion apparaît hypo intense en séquence t1 et iso a hypo intense en T2 avec une prise de contraste annulaire centre par une image hypo intense. Le centre de la lésion devient hyper intense en T2 avec l’apparition du caséum. La partie périphérique du granulome peut apparaître hypo à hyper intense en T2 en fonction du stade d’évolution. L’dème péri lésionnel se présente comme une image hyper intense en T2. Dans notre cas il s’agissait d’une lésion mixte fusiforme, se rehaussant en T1 après injection de gadolinium avec une partie centrale hypo intense. Plusieurs auteurs rapportent l’existence de tuberculomes multiples à l’IRM, de même que la présence simultanée de tuberculome intra-cérébral [9,10,14,18] . YEN [18] recommande la réalisation systématique d’une IRM cérébrale chez des personnes présentant des TIM multiples à cause du caractère très souvent asymptomatique des ces localisations. Le consensus sur le traitement de la TIM n’est pas encore clairement établi [9,13]. CONCLUSION Le TIM, bien qu’étant une affection rare doit être évoqué devant un tableau de compression médullaire lente, surtout dans notre contexte. L’apport des moyens neuroradiologiques modernes dans son diagnostique, l’utilisation des techniques microchirurgicales et l’association d’un traitement anti-tuberculeux adéquat le rendent aujourd’hui potentiellement curable.
 Figure 2  Figure 3
Articles récents
Commentaires récents
Archives
CatégoriesMéta |


 #gallery-3 {
margin: auto;
}
#gallery-3 .gallery-item {
float: left;
margin-top: 10px;
text-align: center;
width: 33%;
}
#gallery-3 img {
border: 2px solid #cfcfcf;
}
#gallery-3 .gallery-caption {
margin-left: 0;
}
/* see gallery_shortcode() in wp-includes/media.php */
#gallery-3 {
margin: auto;
}
#gallery-3 .gallery-item {
float: left;
margin-top: 10px;
text-align: center;
width: 33%;
}
#gallery-3 img {
border: 2px solid #cfcfcf;
}
#gallery-3 .gallery-caption {
margin-left: 0;
}
/* see gallery_shortcode() in wp-includes/media.php */

© 2002-2018 African Journal of Neurological Sciences.
All rights reserved. Terms of use.
Tous droits réservés. Termes d'Utilisation.
ISSN: 1992-2647