
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Example Content AUTOIMMUNE ENCEPHALITIS: A MISSED DIAGNOSTIC AND THERAPEUTIC OPPORTUNITYABSTRACT Encephalitis is a common clinical problem affecting 5 cases per 100 000 population annually. After exclusion of the usual infective causes, a number of cases remain unexplained. It has been observed that many such cases have an autoimmune basis resulting in disruption of synaptic and ion channel function. This diagnosis should be suspected based on subacute onset, short term memory loss, altered mental status or psychiatric symptoms in combination with new focal neurological deficits, new onset seizures, CSF pleocytosis or MRI features suggestive of encephalitis. As this is a treatable condition, with a good prognosis if recognised early, it is important not to miss the diagnosis. Key Words: Autoimmune encephalitis BACKGROUND AND HISTORY The term encephalitis refers to a rapidly progressive encephalopathy (usually in less than 6 weeks) as a result of brain inflammation and has an estimated annual incidence of 5 cases per 100 000 population in a UK cohort.[22, 72] This clinical scenario is quite rightly, in the first instance, considered to have an infectious cause as any delay in the diagnosis and treatment is associated with a high morbidity and mortality. For example, a more than two day delay in treatment initiation of Herpes simplex encephalitis results in a three-fold worse outcome at 6 months.[62] In approximately 50% of cases, the aetiology remains undetermined but with increased awareness many cases previously labelled as idiopathic encephalitis are now being identified to have an autoimmune origin.[47, 77] In view of the initial presentation, many of these patients are mistakenly thought to have a psychiatric disorder resulting in a delay in treatment. The first description of paraneoplastic limbic encephalitis was in 1968 in association with lung, breast and thyroid malignancies.[9] These patients had paraneoplastic anti-Hu or anti-Ma2 antibodies: intracellular antibodies that result in irreversible, cytotoxic T cell responses with a poor prognosis and poor response to therapy. In 2005 Ances et al described 8 patients with limbic encephalitis who, at that time, had unidentified antibodies to neuronal surface proteins.[3] There has since been continued discovery of 1-2 such antibodies per year.[51] These antibodies reversibly disrupt synaptic functions and ion channels and are thus responsive to immunotherapy.[3] Unlike the intracellular neuronal antibodies, these neuronal cell surface antibodies have a more variable association with underlying malignancy (Table 1). In addition, autoimmune encephalitis affects patients of all age groups and detection has diagnostic, prognostic and therapeutic importance. In this review, we discuss the clinical presentation of two of the most common autoimmune encephalopathies supported with case studies. We suggest a practical diagnostic approach to these patients. Table 1: Autoimmune Encephalitis Syndromes
*Limbic encephalitis: subacute confusion, memory disturbance, neuropsychiatric features and seizures. Morvan’s syndrome: cognitive symptoms or seizures, peripheral nerve hyperexcitability and insomnia Abbreviations: AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; CASPR2, contactin-associated protein-like 2; DR2, dopamine receptor 2; DPPX, dipeptidyl-peptidase-like protein-6; GABAR, gamma-amino-butyric acid receptor; GAD,glutamic acid decarboxylase; IgLON5, immunoglobulin-like family member 5; LGI1, leucine-rich, glioma-inactivated 1; mGluR5, metabotropic glutamate receptor 5; NMDAR, N-methyl-D-aspartic acid receptor; PERM, progressive encephalomyelitis with rigidity and myoclonus. METHODS Relevant articles were identified through PubMed searches of articles published in English up to 1 May 2017. Search terms included (alone or in combination) encephalitis, encephalopathy, limbic or autoimmune, which yielded 358 results. Additional articles were identified in references. Priority was given to large case series. ANTI-NMDA RECEPTOR ENCEPHALITIS This disorder is associated with IgG antibodies against the GluN1 subunit of N-methyl D-aspartate (NMDA) receptors.[20] In vitro studies have shown that antibodies bind to NMDA receptors resulting in cross-linking and internalization. The degree of damage correlates with antibody titres and reverses with antibody removal.[31] NMDA encephalitis has been found to be a common cause of encephalitis and the most common form of autoimmune encephalitis.[13] In an English series, 4% of patients with encephalitis were diagnosed with NMDA encephalitis.[21] The California encephalitis cohort identified NMDA encephalitis as being the most common cause of encephalitis, 4 times more common than viral causes, although this cohort was likely influenced by referral bias.[18] An intriguing finding is that in some patients herpes simplex encephalitis may also trigger other autoimmune reactions such as NMDA encephalitis.[15, 27] The earliest reports of NMDA encephalitis were in 2005 and 2007 when Vitaliani and Dalmau described young women with ovarian teratomas presenting with psychiatric abnormalities, movement disorders and central hypoventilation.[14, 76] These descriptions resulted in rapid identification of increasing numbers of cases, with more than 600 cases being described in the literature by 2013.[68] The clinical presentation and association with an underlying tumour varies with age and gender. The median age of presentation is 21 years but has ranged from 8 months to 81 years. Eighty one percent of the patients are female. Associated tumours are present in 38% of patients, predominantly in females between the ages of 12 and 45. Males are more likely to present below the age of 12 or over 45. Ninety seven percent of the associated tumours occur in females, 94% of which are ovarian teratomas. The remainder of tumours are extraovarian teratomas, lung, breast, testicular, ovarian, thymic and pancreatic in origin.[68] These patients typically present in a step wise manner, although steps may occur in any order, all steps do not have to be present and seizures can occur at any stage of disease. In 70% of patients a prodrome may precede the onset of other symptoms by up to 2 weeks. Prodromal symptoms include headache, fever, nausea, diarrhoea or features of an upper respiratory tract infection. Psychiatric symptoms occur next and range from anxiety and insomnia to mania, paranoia, hallucinations and grandiose delusions.[32, 63] In some patients psychiatric symptoms may predominate, resulting in psychiatric admission.[61] Atypical features for schizophrenia, such as pronounced catatonia or associated movement disorders and seizures, should therefore prompt psychiatrists to exclude an underlying autoimmune encephalitis.[70] Short term memory loss is common. Subsequent symptoms include movement disorders, autonomic dysfunction and central hypoventilation. The range of movement disorders is vast including chorea, dystonia, oro-lingual dyskinesias and opisthotonic posturing. The presence of autonomic dysfunction and central hypoventilation often necessitates ventilation and support in an intensive care setting.[32, 63] Monosymptomatic presentations are very rare in all age groups but males more commonly present with partial seizures.[57, 73] Even though the presenting symptoms may vary by age, within 4 weeks the majority of patients have developed a similar cluster of symptomatology.[68] Patients with long-term psychiatric diagnoses have on occasion been found to have NMDA receptor antibodies. The majority of these patients do not have other symptoms described in the syndrome. It is important to note in these patients have IgA or IgM and not IgG antibodies, or antibodies against the GluN2 subunit, and not GluN1 subunit that has been associated with NMDA encephalitis. [6, 28, 41] Four percent of patients have been described to have an overlapping, concurrent or sequential demyelinating syndrome, sometimes with associated Aquaporin 4 antibodies.[67] With treatment 53% of patients improve within 4 weeks and 80% at 24 months. Symptoms often resolve in the reverse order in which they presented. First line treatment consists of tumour removal (if present), steroids, intravenous immunoglobulins and plasmapharesis. Up to 40% of patients do not respond to first line therapy. Second line therapy consists of rituximab or cyclophosphamide (individually or in combination). Approximately 80% of patients who fail first line therapy have a good outcome at 24 months after second line therapy. [68] It is important to keep in mind though that even after improvement of the acute episode there may be persistent deficits in memory, executive function and behavioural disturbances. The condition is associated with a 7% mortality rate and relapses occur in 12-20%. Relapses may be separated from the initial event by months to years, consist of only part of the initial syndrome and are usually less severe than the initial episode.[17, 68] Relapses are also more likely to occur if the presenting episode was not treated or undertreated.[35] Although relapses are less common in those who were treated for an underlying tumour, in the case of a relapse patients should be re-assessed for presence of underlying contralateral or recurrent teratoma.[32] CASE REPORT: NMDA ENCEPHALITIS (IN A BOX) A previously well 19 year old woman was referred to the neurology unit for investigation of a change in her mental state. Five days prior to presentation she reported hearing unfamiliar voices while studying and feeling suspicious of others. Two days later she stopped responding to those around her and subsequently had two generalised tonic clonic seizures. There was no viral prodrome. On examination she was unresponsive to any stimuli and had prominent oromandibular dyskinesias, dystonia and opisthotonic posturing. She required intubation, ventilation and ICU admission. While in ICU she had episodes of supraventricular tachycardia. The patient was commenced on IVI acyclovir and investigated for an encephalitis. The FBC and metabolic panels were normal, HIV ELISA negative and RPR non-reactive. A lumbar puncture showed 8 lymphocytes, 0 polymorphonuclear cells, protein 0.16 g/l, glucose 4.4 mmol/l (plasma 7 mmol/l). The CSF FTA and cryptococcal latex agglutination test were negative, as were the PCR assays for HSV, VZV and CMV. NMDA receptor antibodies were negative in the serum but positive in CSF. The EEG was in keeping with an encephalopathic state with an extreme delta brush pattern. MRI brain was normal. Pelvic ultrasound did not show an underlying ovarian teratoma. The patient did not respond to first line therapy with IV methylprednisone, intravenous immunoglobulins or plasma exchange. She subsequently improved after 5 doses of IV cyclophosphamide. After a three-month admission to ICU she was stepped down to a general ward. She was discharged home two months later with continued outpatient rehabilitation. The patient was continued on oral prednisone and azathioprine and has continued to improve. One year later she is considering resuming her postgraduate studies. VOLTAGE-GATED POTASSIUM CHANNEL ANTIBODIES Voltage-gated potassium channels (VGKC) form part a multiprotein neuronal complex. Prior to 2010 antibodies to the Kv1.1 and Kv1.2 subunits of the Shaker family of VGKC were recognised in association with limbic encephalitis, faciobrachial dystonic seizures, Morvan syndrome and neuromyotonia.[33, 66, 74] Subsequently it has been identified that contactin-associated protein-2 (Caspr2) and leucine-rich, glioma inactivated 1 protein (LGI1) are the true target antigens, each with unique clinical presentations.[34, 43, 71] References to VGKC encephalitis prior to 2010 was most likely referring to an encephalitis secondary to LGI1 antibodies. Antibodies to other extracellular domains of the VGKC are not pathogenic and do not require treatment with immunotherapy.[49] LGI1 ANTIBODIES LGI1 is a protein that interacts with the pre-synaptic ADAM23 and post-synaptic ADAM22 and AMPA receptors, forming a trans-synaptic complex. It is postulated that the antibodies result in reduced AMPA inhibitory behaviour, therefore resulting in neuronal hyperexcitability.[58] LGI1 antibodies result in the second most common form of autoimmune encephalitis, after NMDA.[52] The disorder has a male predominance, with a 2:1 male:female ratio and a median age at presentation of 63 years.[34, 36] The typical presentation is that of faciobrachial dystonic seizures affecting the arm and ipsilateral face, which may precede the onset of limbic encephalitis by up to 26 days.[36] These seizures last about 3 seconds each and may occur a median of 50 times per day. Identification of the disorder at this stage is important as early treatment may prevent the rest of syndrome from developing and improves the prognosis.[38] Limbic encephalitis then develops, which consists of subacute onset confusion, short-term memory disturbance and personality changes.[34] Patients may then go on to develop generalised tonic clonic seizures, focal seizures with impaired awareness of temporal lobe origin or focal seizures with associated piloerection.[34, 74, 79] Presentation in non-convulsive status is not uncommon. As these seizures do not respond to anticonvulsants but to immunosuppressive therapy the identification of the disorder is very important.[36] A unique feature of LGI1 encephalitis is hyponatraemia, which occurs in 60-88% of cases. This is thought to be due to LGI1 antibodies binding to antidiuretic hormone-secreting neurones.[37] In the majority of patients, there is no tumour association. Prognostically, 71% of patients are seizure free with monthly IV MTP but only 33% had normalization of memory.[54] Rapid identification and treatment of this disorder is important as a delay in institution of immunotherapy has been associated hippocampal atrophy and poorer long-term memory outcome.[16] Therefore, despite patients with LGI1 encephalitis having a more rapid response to treatment than those with NMDA encephalitis it is likely that they have a poorer long-term recovery.[51] Reported mortality is 6% and relapses occur in 15%.[34, 43] CASPR2 ANTIBODIES Disease associated with Caspr2 antibodies is more diverse, consisting of central and peripheral nervous system involvement. Limbic encephalitis (42%) and Morvan syndrome (29%) are the most frequent clinical syndromes and develop over an average of 4 months.[71] Morvan syndrome refers to a combination of cognitive symptoms or seizures, peripheral nerve hyperexcitability and insomnia. CASE REPORT: LGI1 ENCEPHALITIS (IN A BOX) A 65 year old man with a background history of diabetes and hypertension presented in April 2014 with a first episode generalised tonic clonic seizure lasting 10 minutes. He had no further neurological complaints and a normal neurological examination. He also had features of a right middle lobe pneumonia and cholangitis. The serum Na was 117 mmol/L with a urinary Na of 75 mEq/L, urine osmolality of 536 mOsm/kg and serum osmolality of 269 mOsm/kg. This was interpreted as syndrome of inappropriate ADH on the basis of underlying septicaemia, complicated by a seizure. A CT brain was normal. The underlying septicaemia was treated and he was commenced on fludrocortisone tablets and water restriction. DIAGNOSTIC EVALUATION An encephalitis should be suspected in any patient presenting with combinations of subacute onset (in the last 3 months) of confusion, behavioural change, change in mood or seizures. Initially, a viral cause is presumed and treatment should be commenced pending results of initial investigations (Table 2).[72] It should be kept in mind that despite high assay sensitivity and specificity, CSF HSV PCR may be falsely negative if performed in the first 24 hours of the illness and should therefore be repeated if clinically suspected.[78] Table 2: Initial Evaluation of a Patient Presenting with Encephalitis
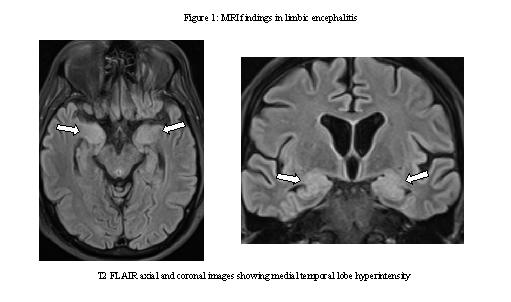
Abbreviations: EEG, electroencephalogram; FTA, fluorescent treponemal antibody absorption; TPHA, treponemal pallidum haemagglutination assay; WCC, white cell count The majority of patients with autoimmune encephalitis have an abnormal lumbar puncture, with the exception being patients with LGI1 antibodies (normal in 59%).[39, 51] The most common findings are pleocytosis of less than 100 cells per L in 80%, mild-moderate increase of CSF protein in 30% and the presence of oligoclonal bands in 60%.[51] The EEG is abnormal in most patients, and usually shows slow frontal or temporal activity, with or without associated epileptiform discharges.[40] Although the EEG is a useful test to rule out alternative causes of encephalopathy the only antibody specific finding is that of the extreme delta brush pattern in 30% of patients with NMDA receptor encephalitis.[64] Typical MRI findings include T2/FLAIR hyperintensities in one or both medial temporal lobes (Fig. 1). These findings are however not specific, and may also occur in herpes encephalitis and in seizures of temporal lobe onset.[50, 59] Similarly, a normal MRI doesn’t exclude the diagnosis, as it may be normal in 66% with NMDA encephalitis [68] and 46% with LGI1 encephalitis.[36] However, even in patients with normal appearing MRI scans, medial temporal lobe hypermetabolism has been identified on FDG-PET imaging.[4, 30] Due to the overlap in symptomatology, antibody testing is available as a commercial panel that includes antibodies against Caspr2, LGI1, NMDA, AMPA and GABAb. Testing against other antibodies is only performed in research laboratories. Detection of CSF antibodies to cell surface antigens strongly supports the diagnosis whilst the significance of antibodies in serum only has less clear significance.[60] In a study looking at paired CSF and serum samples in NMDA encephalitis, up to 14% of cases were serum negative but CSF positive. Therefore, when testing for neuronal surface antibodies it is advisable to do so in both the CSF and serum for a number of reasons: As early treatment improves outcome,[3, 7, 74] there has been a recent shift in guidelines towards earlier recognition and treatment of autoimmune encephalitides.[25] These guidelines are less dependent on results of antibody tests, as these may be negative in 10%, may not be accessible in all instances or may take weeks to obtain.[24] Despite these limitations the identification of specific antibodies still has value as a prognostic tool, guides the search for associated tumours and provides a definitive diagnosis. In cases with a strong clinical suspicion of autoimmune encephalitis repeat testing of both CSF and serum samples should be performed as the finding of delayed seropositivity has been described.[65] Therapy with intravenous immunoglobulin may make result in false negative results, but this finding is less common in the CSF than serum. It is however suggested that in cases where patients fulfil the criteria for probable autoimmune encephalitis, and other causes have been excluded, treatment should be offered regardless of antibody status.[25] Possible autoimmune encephalitis should be suspected based on the presence of the following three criteria:[25] (put in a box) 1. Subacute onset (within the last 3 months) of short term memory loss, altered mental status (personality change, lethargy or altered level of consciousness) or psychiatric symptoms 2. At least one of: a. new focal neurological deficits b. new onset seizures c. CSF pleocytosis (white cell count of more than 5 cells/mm3) d. MRI features suggestive of encephalitis 3. Exclusion of other causes Therefore in the presence of these features, and the exclusion of abnormalities in the initial evaluation, patients should be referred for further neurological workup and management.
TREATMENT Intracellular, paraneoplastic autoimmune encephalitides, with anti-Hu/Ma-2 or GAD antibodies are managed by treatment of the underlying malignancy. The response to therapy and prognosis in these patients is generally poor due to the irreversible T cell driven cytotoxicity.[44] In those with antibodies to cell surface antigens, however it has been established that early treatment with immunomodulatory therapy improves outcome, and may halt progression of the syndrome.[7, 36, 74] Published experience in treatment of NMDA and LGI1 encephalitis has largely determined the treatment approach in these patients.[34, 43, 68] There are however no specific guidelines for the treatment of autoimmune encephalitis, nor therapies that specifically target antibodies. Based on experience first line therapy consists of tumour removal (if indicated), steroids, intravenous immunoglobulins or plasma exchange. Patients who have a tumour have a worse outcome and more relapses if not resected.[32] If there is no response within 4 weeks therapy may be escalated to rituximab or cyclophosphamide. There has been a recent move towards the earlier use (or first line use) of second line therapy as this may result in a lower relapse rate with good tolerability.[68] The rationale is that these agents target antibody production by plasmablasts rather than neutralising or eliminating already formed systemic antibodies.[53] It is currently unknown how long treatment should be continued for patients with these disorders, thus individualised patient care decisions should be made. Ongoing multidisciplinary care is of utmost importance in these patients and team members will vary during the disease course. It is important to note that these are disorders requiring high degrees of therapeutic commitment as average length of hospital admission is 3 months (NMDA encephalitis) with a longer period of rehabilitation thereafter.[51] It is therefore necessary to assemble a personalised team of specialists potentially consisting of neurologists, intensivists, neuropsychologists, psychiatrists and rehabilitation personnel in order to optimise patient outcome.[56] TUMOUR SCREENING Unlike encephalitides with antibodies against intracellular proteins (anti-Hu/Ma2), the association with an underlying malignancy is variable in patients with neuronal cell surface antibodies. The need for tumour screening should therefore be guided by the identified antibody together with the neurological syndrome, age and gender of the patient (see table 1). Syndromes with a low likelihood of associated malignancy may therefore not justify ongoing evaluation. In cases with high tumour associations however, if initial screening is negative repeat this should be repeated after 3-6 months and then 6 monthly for up to 4 years. Appropriate screening modalities for the thorax are a CT chest followed by FDG/PET, breast screening with a mammogram and screening for ovarian tumours with a transvaginal ultrasound followed by a CT pelvis. Screening with tumour markers alone is not reliable. [69] CONCLUSION With increased awareness autoimmune encephalitis has become one of the most frequent causes of encephalitis. This is a treatable condition affecting all age groups, and at times preferentially young individuals. Despite the poor prognosis in cases where diagnosis is delayed, with treatment a good outcome can be expected in 70-80% of individuals.[47] Thus, an increased awareness with prompt identification and treatment of these patients is paramount. ACKNOWLEDGEMENTS Dr B Bhagwan and Dr S Marais kindly allowed us to use the case reports of their patients with LGI1 and NMDA receptor encephalitides respectively. CONFLICT OF INTEREST None AUTHOR CONTRIBUTIONS AIB conceived the design and undertook critical analysis of the paper. IR participated in concept design, undertook the literature search and wrote the initial draft of the paper. The final manuscript was approved by both authors. FUNDING SOURCES None REFERENCES
Articles récents
Commentaires récents
Archives
CatégoriesMéta |

© 2002-2018 African Journal of Neurological Sciences.
All rights reserved. Terms of use.
Tous droits réservés. Termes d'Utilisation.
ISSN: 1992-2647