
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
INTRODUCTION Les méningiomes intracrâniens sont des tumeurs qui se développent aux dépens des cellules arachnoïdiennes des méninges qui recouvrent l’encéphale ; ils ne correspondent donc pas, stricto sensu, à des tumeurs cérébrales. Cependant, leur croissance endocrânienne et les conséquences neurologiques rendent compte du fait qu’ils soient classés parmi les tumeurs cérébrales [5]. Les méningiomes sont les tumeurs primitives intracrâniennes les plus fréquentes chez l’adulte. Ils sont de degré histologique variable [11,12]. Le choix de la modalité de prise en charge chirurgicale dépend des facteurs liés au patient (âge, état général, comorbidités) et au plateau technique. Le but de la chirurgie est d’aboutir à une exérèse la plus complète possible, et de permettre une analyse histologique. En effet, les patients ayant bénéficié d’une exérèse complète présentent un taux de récidive sur 10 ans de l’ordre de 9%, comparés aux patients chez lesquels l’exérèse a été incomplète [16]. En Afrique subsaharienne, l’amélioration de l’accès aux moyens de neuro-imagerie a contribué à faciliter le diagnostic des tumeurs intracrâniennes. En Côte d’Ivoire, les méningiomes constituent 33,43% des tumeurs intracrâniennes, suivis des adénomes hypophysaires (20%) et des astrocytomes (10,30%) [14]. À Brazzaville (République du Congo), les méningiomes représentent 39,62% des tumeurs intracrâniennes, suivis des gliomes de haut grade (35,84%). Mais, leur prise en charge est marquée par une mortalité globale estimée à 38,33%, avec une mortalité post-opératoire à 10% [9,10]. Dans notre contexte, aucune étude n’a été menée concernant la prise en charge des méningiomes qui constituent les tumeurs intracrâniennes les plus fréquentes. Le but de notre étude était de décrire la prise en charge neurochirurgicale des méningiomes intracrâniens, afin de contribuer à réduire le taux de mortalité lié aux tumeurs crânio-encéphaliques. PATIENTS ET MÉTHODE Nous avons réalisé une étude descriptive, à recueil de données rétrospective, du 1e janvier 2014 au 31 décembre 2017 (48 mois). Cette étude a été menée au sein de l’unité neurochirurgicale du service de Chirurgie Polyvalente du Centre Hospitalier Universitaire (CHU) de Brazzaville, qui compte quatre neurochirurgiens et constitue au plan national, la structure de référence pour la prise en charge des affections neurochirurgicales. Nous avons sélectionné tous les patients chez lesquels le diagnostic de méningiome était évoqué et exclu les cas avec données incomplètes. Ces patients étaient admis en provenance du service des urgences ou de la consultation programmée de neurochirurgie. Le diagnostic de méningiome était retenu à partir des données de la tomodensitométrie (TDM) seule et ou de l’imagerie par résonance magnétique (IRM), avec une lésion d’allure extra-axiale, adjacente à l’os du crâne, avec une prise de contraste diffuse de la tumeur et de la méninge adjacente, pouvant donner l’aspect « en queue de comète ». Les lésions étaient classées en méningiomes supra et sous-tentoriels. Ceux supra-tentoriels étaient également classés en méningiomes de la convexité (proches du sinus sagittal supérieur ou non), et en méningiomes de la base du crâne (étages antérieur et moyen). L’indication de chirurgie était retenue lorsque le score de l’American Society of Anesthesiologists (ASA) [7] était compris entre 1 et 3, avec des conditions d’accès et d’exérèse sans dommage fonctionnel et risque de décès majeurs en per opératoire. Le traitement médical péri-opératoire incluait les corticostéroïdes (1 à 2 mg/kg par jour), la prophylaxie antiépileptique avec le phénobarbital (3mg/kg par jour) ou le valproate de sodium (20 à 30 mg/kg par jour), en cas de méningiome supra-tentoriel ou en cas d’hydrocéphalie (liée à un méningiome sous-tentoriel). Le mannitol à la dose de 0,5 à 1g/kg était utilisé pour ses propriétés osmotiques. L’énoxaparine à la dose de 0,4 ml/jour en injection sous-cutanée était systématique chez les patients alités et 24 heures après la chirurgie. La prescription des inhibiteurs de la pompe à protons était systématique. Les moyens chirurgicaux étaient constitués par la dérivation ventriculo-péritonéale (DVP) et l’abord direct de la lésion pour exérèse. L’indication d’une DVP était discutée en cas d’hydrocéphalie, selon le statut clinique du patient, la topographie du méningiome et la possibilité ou non d’obtenir une résolution de l’hydrocéphalie avec l’exérèse de la lésion seule. Les moyens utilisés pour la chirurgie d’exérèse étaient : la têtière à pointes de Mayfield, le microscope opératoire et la coagulation bipolaire. La qualité de l’exérèse était appréciée en utilisant la classification de Simpson [18]. En cas d’exérèse complète emportant la dure-mère, une plastie de fermeture était réalisée avec la galéa et de la colle biologique. Dans les situations d’exérèse avec craniectomie perdue (os infiltré), une cranioplastie de remplacement était organisée dans un second temps chirurgical, à distance de la première intervention. En cas de suites opératoires simples, le patient était hospitalisé pendant quatre à sept jours, la TDM de contrôle était proposée deux à trois mois après la chirurgie. Les patients perdus de vue au contrôle clinique post-opératoire étaient contactés par téléphone. Les paramètres évalués étaient cliniques, radiologiques, histologiques, thérapeutiques et évolutifs. La collecte des données a été réalisée à partir d’un registre des hospitalisations au sein de l’unité, des dossiers médicaux, et par appel téléphonique. Ces données ont été traitées avec le logiciel Numbers de Macintosh 5.3 2008-2018 Apple Inc. RÉSULTATS Population d’étude Durant la période d’étude, 119 cas de tumeurs crânio-encéphaliques ont été enregistrés, dont 47 méningiomes, soit une fréquence de 39,5%. Trois cas ont été exclus de l’étude pour insuffisance de données. Ainsi, 44 cas de méningiomes (36,9%) ont constitué notre série. Caractéristiques anthropométriques L’âge moyen était de 57,11 ± 15 ans, avec des extrêmes de 26 et 84 ans. Nous avons identifié 12 hommes contre 32 femmes, soit un sex ratio de 0,37. Aspects cliniques La médiane pour le délai d’admission était de 39 mois, avec des extrêmes de 2 et 96 mois. Les antécédents médicaux des patients étaient caractérisés par l’hypertension artérielle seule dans sept cas (16%), le diabète seul dans deux cas (4,5%) et l’association des deux dans cinq cas (11,4%). Le tableau I représente la répartition des patients selon les signes cliniques à l’admission. La médiane pour le score de Glasgow était de 15, avec des extrêmes de 6 et 15. Trois patients avaient un score de Glasgow inférieur ou égal à 8. Le score ASA médian était de 1, avec des extrêmes de 1 et 6. Treize patients (29,5%) avaient un score ASA supérieur ou égal à 4. Données neuroradiologiques La TDM et ou l’IRM ont été réalisées chez tous les patients de la série. La TDM a été réalisée seule dans 26 cas (59,1%), l’IRM seule dans quatre cas (9,1%), ces deux examens dans 14 cas (31,8%). Les figures 1 et 2 montrent deux cas de méningiomes : le premier est un méningiome de la convexité occipitale droite, falco-sinusien, le second est un méningiome à cheval sur les étages moyen et postérieur de la base du crâne à gauche, exerçant un effet de masse sur le tronc encéphalique. Le tableau II représente la répartition des patients selon la topographie des méningiomes. La taille moyenne des méningiomes était de cinq centimètres, avec des extrêmes de 1,2 et 8. Le tableau III représente la répartition des patients selon la taille du méningiome. L’œdème péri-lésionnel était identifié dans 31% des cas (70,4%) : il était de taille inférieure à celle de la tumeur dans sept cas (15,9%), égale à celle de la tumeur dans sept cas (15,9%) et supérieure à la tumeur dans 17 cas (38,6%). Traitement chirurgical Dans notre série, 25 patients (56,8%) ont été opérés. Deux patients (8%) ont été traités par DVP pour hydrocéphalie obstructive, avec un méningiome du foramen ovale. Les 23 autres patients ont subi une chirurgie d’exérèse Simpson I dans sept cas (30,4%), II dans quatre cas (17,4%), III dans cinq cas (21,7%), IV dans quatre cas (17,4%) et V dans un cas (4,3%). Deux patients sont décédés en début d’intervention, par hémorragie, durant la confection du volet osseux. Les cas d’exérèse tumorale complète de type Simpson I concernaient un abord frontal dans trois cas, pariétal dans deux cas, orbitaire et ptérional dans un cas respectivement. Parmi ces patients, un cas a bénéficié d’une cranioplastie à distance. L’hémorragie per opératoire était jugée importante, avec recours à une transfusion sanguine dans six cas, parmi lesquels les deux décès par hémorragie durant la confection du volet osseux. Le diagnostic histologique n’a été identifié que dans sept cas. Il s’agissait tous de méningiomes endothéliomateux. Aspects évolutifs L’évolution post-opératoire a été favorable dans 15 cas (60%), marquée par une amélioration clinique et une qualité d’exérèse satisfaisante. Seul un patient a été vu à deux ans de la chirurgie, sans récidive tumorale à la TDM et à l’IRM avec injection de produit de contraste. Aucun autre patient n’a pu réaliser une TDM au-delà du premier contrôle réalisé au cours du premier semestre post-opératoire. Trois patients (12%) ont eu une évolution post-opératoire initialement stationnaire. Ils avaient été opérés avec une exérèse tumorale de type Simpson IV (deux cas) et V (un cas). À deux ans d’évolution, un patient a été perdu de vue, un autre a développé une infection pulmonaire à distance et son état clinique a été compliqué par une perte progressive de l’autonomie. Deux patients porteurs d’hydrocéphalie obstructive avec un méningiome jugé inaccessible par la chirurgie (foramen ovale) ont eu une évolution marquée par une dégradation progressive de l’état neurologique à deux ans de la chirurgie. L’imagerie avait dans ces cas montré une extension sous-tentorielle et compressive sur le tronc cérébral. Cinq patients (11,3%) ont exprimé un refus de la chirurgie. Ils étaient ensuite perdus de vue. Dix sept patients (38,6%) sont décédés. Parmi ces cas, 13 patients n’ont pas été opérés, et les quatre cas opérés (16%) étaient décédés suite à une hémorragie per opératoire dans deux cas, et à une poussée d’œdème post opératoire malgré la prise en charge en réanimation dans deux cas. DISCUSSION Notre étude a été rétrospective, ce qui constitue une limite dans la qualité du recueil des données. La durée d’étude au prorata du nombre de cas enregistré n’est pas suffisante pour procéder à une étude analytique sur les facteurs de morbi-mortalité et la pertinence des moyens à proposer afin d’obtenir un réel impact sur la mortalité post opératoire. L’insuffisance du diagnostic anatomo-pathologique constitue également une limite considérable, eu égard à la nécessité de classer les patients selon la sévérité histologique et de proposer des modalités thérapeutiques complémentaires à la chirurgie. Néanmoins, la description globale de la série a permis d’identifier les symptômes révélateurs de tumeurs cérébrales en général dans notre contexte, de décrire les conditions de sélection des cas pour la chirurgie, avec un retard diagnostique qui contribue probablement à dégrader l’état général des patients et à expliquer le nombre de cas non opérés qui est de l’ordre de 40%. Épidémiologie La fréquence des méningiomes dans notre série a été estimée à 39,5%. En effet, les méningiomes intracrâniens constituent les tumeurs cérébrales les plus fréquentes à Brazzaville, suivis des gliomes de haut grade, des épendymomes et des métastases [10]. En Côte d’Ivoire, leur fréquence est estimée à 33,43% [14]. À Enugu (Nigeria), cette fréquence est de 23,8% [12] et à Lomé (Togo), elle est de 45,6% [8]. Une augmentation de l’incidence des méningiomes est rapportée ces dix dernières années dans de nombreuses études. Il a été suggéré qu’il pourrait entre autres s’agir de la conséquence du vieillissement de la population combinée à l’amélioration de l’accès aux ressources de santé et aux procédures diagnostiques [2]. En outre, les données de la littérature suggèrent la participation d’un facteur racial, marqué par une fréquence plus importante des méningiomes chez les individus de race noire [20]. L’âge moyen dans notre série était de 57.11 ± 15 ans. Il varie entre 40 et 50 ans dans les séries subsahariennes [12,14,19]. La prédominance féminine des méningiomes a été identifiée par Harvey Cushing il y a un siècle environ. Elle suggère l’implication des facteurs hormonaux et génétiques [2]. Dans notre série, le sex ratio était de 0,37. Diagnostic Le délai médian entre le début des symptômes et l’admission dans notre étude était de 39 mois, avec des extrêmes de 2 et 96 mois. En Côte d’Ivoire [14], le délai moyen pour le diagnostic était de 22 mois ; ce délai était raccourci à 6 mois dans l’étude de Thiam et al. [19] à Dakar au Sénégal. Les méningiomes constituent des tumeurs à croissance majoritairement lente, avec une symptomatologie initiale le plus souvent insidieuse [16]. Au plan clinique, l’hypertension intracrânienne (HTIC) était retrouvée dans 81,8% des cas. Nous avons inclus les cas de céphalées persistantes et inhabituelles, avec ou sans œdème papillaire, comme liés à l’HTIC, lorsqu’ aucune autre raison évidente ne pouvait les expliquer. N’dri Oka et al. [14] en Côte d’Ivoire avaient retrouvé des céphalées isolées dans 68,42% des cas. Les céphalées d’apparition et d’évolution progressive sont communément associées à l’HTIC, reflétant le rythme de croissance lentement progressif des méningiomes [6]. Les méningiomes de la convexité étaient les plus fréquents dans notre série (79,5%), avec la localisation frontale en premier (43,2%). Cette tendance est rapportée dans la littérature, sans tenir compte de la proximité ou non du sinus sagittal supérieur [6,16]. Nos résultats sont similaires à ceux de N’dri Oka et al. [14] en Côte d’Ivoire qui avaient trouvé une fréquence de 47,36%. À Enugu (Nigéria), Mezue et al. [12] avaient identifié les méningiomes de la gouttière olfactive dans 26,5% des cas, suivis par les méningiomes de la convexité dans 23,5% des cas, et ceux para-sagittaux dans 17,7% des cas. Thiam et al. [19] à Dakar (Sénégal), avaient identifié les méningiomes de la convexité en premier, dans 48% des cas. La localisation intraventriculaire n’a pas été trouvée dans notre série. Dans une étude rétrospective sur 80 cas de méningiomes de taille supérieure ou égale à 5 centimètres, Narayan et al. [13] en Louisiane (États-Unis d’Amérique) avaient trouvé une taille moyenne de 56.4 ± 4 mm avec des extrêmes de 50 mm et 84 mm. La résection des volumineux méningiomes est considérée comme un challenge, du fait de leur fréquente hypervascularisation, de l’œdème péritumoral associé à une HTIC, et des rapports étroits avec des structures neurovasculaires dont l’identification n’est pas toujours aisée [4]. En Côte d’Ivoire, N’dri Oka et al. [14] avaient trouvé 66% des patients avec une taille de méningiome comprise entre 3 et 6 cm, et 23,96% avec une taille supérieure à 6 cm. Dans notre série, la taille moyenne était de 5 cm (extrêmes de 1,2 et 8 cm). Dans notre étude, l’œdème péri-lésionnel était observé dans 31 cas (70,4%), avec un rapport à la taille du méningiome dont la distribution était la suivante : inférieur (15,9%), égal (15,9%) et supérieur (38,6%). N’dri Oka et al. [14] avaient trouvé un œdème péri-lésionnel dans 41,05% des cas. L’œdème associé aux méningiomes a une origine vasogénique ; elle est en rapport avec la production d’un facteur de croissance vasculaire endothélial par la tumeur, le tout étant responsable d’un effet de masse et d’une infiltration veineuse facilitée par la congestion vasculaire. Ces éléments seraient suggestifs d’un potentiel de récurrence de certains méningiomes [17]. Traitement chirurgical et évolution Le choix du type de traitement des méningiomes tient compte de facteurs liés aux patients (état général, comorbidités), et aux types de traitement disponibles. Le but de la chirurgie est d’obtenir une exérèse complète [16]. Dans notre série, la médiane pour le score de Glasgow était de 15, mais 29,5% des patients avaient un score ASA défavorable, ce qui justifie qu’ils aient été récusés pour la chirurgie. Les méningiomes de la convexité sont généralement considérés comme plus accessibles à la chirurgie. Cependant, la proximité avec le sinus sagittal supérieur et ou le torcular pose le problème de la chirurgie des méningiomes proches de la ligne médiane. Les principes de la chirurgie des méningiomes de la base reposent sur une craniectomie suffisante pour minimiser les risques de lésions parenchymateuses au cours de l’exérèse, un suivi des plans arachnoïdiens, l’identification et le respect des rapports vasculaires, et la résection la plus complète possible lors de la première intervention [3]. Dans notre série, parmi les sept cas de chirurgie avec exérèse complète Simpson I, cinq correspondaient à une chirurgie de la convexité (frontale et pariétale). Dans la littérature, malgré une exérèse macroscopiquement complète, jusqu’à 20% des méningiomes récidivent dans les 10 ans après la chirurgie ; la récidive à 10 ans pour les méningiomes opérés avec exérèse incomplète est de plus de 80%. Les tumeurs de la base du crâne sont d’exérèse difficile, le plus souvent incomplète, du fait de la proximité avec des structures vasculaires ou nerveuses hautement fonctionnelles [5]. Le recul de deux ans dans notre étude est insuffisant pour apprécier le taux de récidive de ces méningiomes. Par ailleurs, le suivi des patients après chirurgie est difficile dans notre contexte, à cause des conditions socio-financières, qui constituent une limite quant à la réalisation des contrôles radiologiques. Le but de la chirurgie dans les méningiomes para-sagittaux est d’obtenir une exérèse la plus complète possible, sans complications, en préservant au mieux la circulation veineuse cérébrale. En cas de reliquat tumoral de petite taille et d’exérèse difficile, la radiochirurgie stéréotaxique constitue un traitement complémentaire valable, sans pour autant constituer une alternative à la chirurgie d’exérèse réalisée dans des conditions optimales [15]. Dans notre contexte, le but de la chirurgie était de réduire au maximum la taille de la tumeur, et de proposer une prise en charge complémentaire en fonction des données histologiques et du contrôle radiologique. La radiothérapie pour le névraxe n’était pas disponible au Congo durant la période d’étude. L’insuffisance de recul dans notre étude est les difficultés à obtenir un contrôle radiologique n’ont pas permis d’évaluer les indications de radiothérapie après chirurgie. Dans notre série, 17 patients (38,6%) sont décédés, parmi lesquels, quatre dans le groupe des patients opérés (16%). En Côte d’Ivoire [14], cette mortalité était de 12,63%, et au Sénégal [19], elle était de 10,25%. Le pronostic des méningiomes dépend des facteurs comme le grade histologique de la tumeur, la qualité de l’exérèse et les possibilités de traitement complémentaires [1]. CONCLUSION Les méningiomes intracrâniens constituent les tumeurs les plus fréquentes en pratique neurochirurgicale dans notre contexte. L’HTIC est le principal mode de révélation de ces tumeurs. Le diagnostic est facilité par l’accessibilité à la TDM et à l’IRM. La chirurgie demeure la principale procédure thérapeutique, notamment lorsque l’exérèse tumorale est complète. L’évolution dépend du grade histologique et des possibilités de traitement complémentaires à la chirurgie d’exérèse. Dans notre étude, les méningiomes de la convexité avaient le meilleur pronostic à court et moyen termes. La prise en charge optimale des méningiomes de la base du crâne nécessite un perfectionnement de notre pratique chirurgicale pour les lésions de cette zone. La disponibilité des données histologiques et la mise en place des moyens complémentaires comme la radiothérapie peuvent contribuer à améliorer la prise en charge des méningiomes de la base du crâne, en fonction de la qualité de l’exérèse chirurgicale initiale.
Tableau I. caractéristiques cliniques des patients de la série.
*HTIC (Hypertension intracrânienne) : avec œdème papillaire dans 2 cas, avec cécité dans 3 cas. **Déficit neurologique focal : syndrome frontal (6 cas), syndrome opto-chiasmatique (2 cas), paralysie faciale périphérique (2 cas).
Tableau II. Topographie des méningiomes dans notre série.
Tableau III. Répartition des patients selon la taille du méningiome en centimètres (cm).
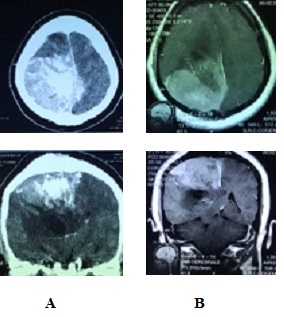 Figure 1: Meningiome falco-sinusien occipital droit.
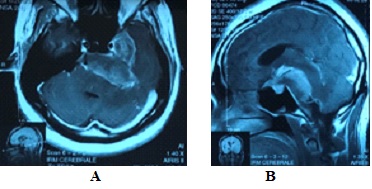 Figure 2: Méningiome de la base, étendu de l’étage moyen à la fosse crânienne postérieure. INTRODUCTION GBS is a heterogeneous group of related disorders, which shares a common pathogenesis and several clinical features, including: an infection history, monophasic disease course and symmetrical cranial or limb weakness (10). His PCB variant is a rare entity not known by most neurologists. Therefore, patients presenting with PCB are initially misdiagnosed as having diphtheria, brainstem stroke, myasthenia gravis (MG) or botulism (3,7,11). A history of campylobacter jejuni diarrhea is frequent. The study of nerve conduction suggests, often, an axonal more than a demyelinating mechanism. Nevertheless, an EMG done early can be normal without eliminating the diagnosis (3). PCB syndrome is frequently associated with the presence of antigangliosides anti-GT1a and possibly anti-GQ1b. Although PCB variant of GBS is rare, it should always be considered in patients presenting to the emergency room with symptoms and signs suggesting progressive brainstem dysfunction such as atypical multiple cranial nerve palsy with normal cerebral MRI (10). We report a case of PCB syndrome in a 30-year-old male; presenting with descending oculopharyngeal and cervicobrachial impairment who was initially misdiagnosed as having a brainstem lesion, botulism or myasthenia gravis, thus highlighting the diagnostic difficulty in an atypical presentation of GBS. PATIENT AND OBSERVATION A 30-year-old male patient presented at the emergency room with horizontal diplopia and upper limbs weakness, which occurred 10 days after an acute diarrhea. Seven days later, he developed dysarthria, dysphagia with outflow of liquids through the nose and chewing disorder. He was admitted to our department at day 14. The initial examination found a conscious patient without any sign of meningeal irritation. He had dysarthria, dysphonia, severe dysphagia for solid and liquid food, bilateral facial palsy, diplopia with ophthalmoplegia, (Fig 1A, B, C, D) and preserved photomotor reflex without any sensory disturbance. Manual muscle testing revealed a -3/5 weakness in the cervical muscles and a -4/5 weakness in the shoulder and proximal upper limbs muscles; muscles of lower limbs were normal. At deep tendon reflexes testing, we found hyporeflexia in upper limbs, normal in lower limbs. In addition, he had dysautonomic disorders such as constipation, tachycardia and hypersudation. Brain MRI, otorhinolaryngological examination with cavum exploration did not show any abnormal lesion. The prostigmine test was normal as was the EMG performed at day 10. The anti-RACH and anti-MUSK antibodies were negative. CSF analysis found no sign of infection nor albuminocytological dissociation. The repetitive nerve stimulation study (RNS) did not show a significant decrement of the compound muscle action potentials (CMAPs). There was an elongation of distal latencies with a decrease in the amplitude of motor potential at the EMG performed on day 18 (Fig 2). Determination of antiganglioside antibodies revealed positive Anti-sulfatide IgM antibodies. The patient was diagnosed having PCB variant of GBS, and we treated him with immunoglobulin. He received daily intravenous immunoglobulin, 400mg/kg/day, for 5 days. His symptoms improved a week afterwards with complete recovery in 6 weeks (Fig 3A, B, C, D). DISCUSSION PCB syndrome was first described as a regional variant of GBS in three cases reported by Ropper in1986. It’s a rapidly progressive oropharyngeal, neck and shoulder weakness, in the absence of sensory disturbance and with relative sparing of the lower limbs (6,9,11). In many clinical reports, the presence of additional features indicates an overlap with other GBS variants: ataxia with ophthalmoplegia suggests an overlap with Fischer syndrome (FS) (5,12); and if in addition consciousness is impaired, an overlap with Bickerstaff brainstem encephalitis (BBE) is suggested (9,11). The existence of PCB with overlapping FS and BBE provides clinical evidence that PCB and these conditions form a continuous spectrum (6). Many clinical forms of PCB are described in the literature; patients presenting with rapidly progressive oropharyngeal and cervicobrachial weakness associated with hyporeflexia or areflexia in the absence of ophthalmoplegia or leg weakness are defined as ‘pure’ PCB (6,11). On the other hand, those who also displayed normal or exaggerated reflexes, which is apparent in about 10% of GBS are defined as having ‘PCB with preserved muscle stretch reflexes’ (3,6,9,11). Incomplete forms of PCB syndrome have also been described: patients without arm and neck weakness have « acute oropharyngeal palsy »; patients without pharyngeal palsy have « acute cervicobrachial weakness », « fulminant PCB weakness » is when patient develop early and severe leg weakness (9). The findings of a prospective study of more than 250 patients with GBS done at the Massachusetts General Hospital showed that FS (5%) was the most frequent GBS variant with PCB (3%) as the second (9,11). In the largest case series which examined 100 patients with PCB and its overlap syndromes in Japan, a history of upper respiratory tract infectious symptoms and diarrhea were observed in 71% and 30% of patients respectively. Similar to GBS, 31% had serological evidence of Campylobacter jejuni infection (6,11). There are also non-infective associations reported: various vaccines, autoimmune diseases, immunosuppressive drugs, and surgery (10). Our patient had a history of diarrhea 10 days before symptoms’ onset, but Campylobacter jejuni serology wasn’t performed. In patients with PCB syndrome, some leg weakness may be present, but oropharyngeal, neck and arm weakness should be more prominent (9). Some patients may also develop additional facial weakness or ophthalmoplegia (6,9,12). In the 100 case series of PCB of nagashima and al; the most frequent initial symptom was arm weakness (29.0%), followed by dysphagia (17.0%) and diplopia (17.0%) (3,6). Predominant arm weakness was proximal in 47.0% and distal in 28.0%. Autonomic dysfunction occurred in 20.0% of cases (6). Our patient consulted initially for diplopia otherwise he had dysautonomia and bilateral facial weakness. The diagnosis of PCB can be challenging in early disease stage. Patients are initially misdiagnosed as having brainstem stroke, MG or botulism (3,4,9,11), diphtheria, brainstem tumor, neurobehcet disease, multiple sclerosis (6,10). Otherwise, some case reports have described the comorbidity of MG and GBS (8). Brainstem ischemia should be considered if symptoms start abruptly, while fluctuations in weakness or obvious fatigability are more suggestive of MG. Botulism is also characterised by ptosis, internal and external ophthalmoplegia, bulbofacial weakness and symmetrical descending flaccid paralysis (3,11). However, MG and botulism can usually be excluded in the presence of sensory deficits or absent reflexes; if doubt persists low and high frequency repetitive nerve stimulation and triple stimulation technique can be used to assess patients with MG and botulism respectively; particularly in the early stages when electrodiagnostic criteria for axonal GBS do not fit the clinical criteria (7). Differential diagnoses were discussed in our case; mainly brainstem stroke, MG or botulism; because of atypical presentation. Therefore, many investigations have been performed uselessly. The neurophysiological findings in PCB are axonal rather than demyelinating. It can be normal in early disease stage. In that case, repeated testing after few days is advised (3,11). When comparing anti-ganglioside antibody–mediated GBS, PCB is considered a nodo-paranodopathy, because reversible conduction failure has been detected by serial conduction studies in the distal and intermediate motor nerve segments (2,7). The most important early investigation is brain MRI to exclude brainstem ischemia, inflammation or brain tumours, which may also involve the skull base or meninges (3,10,11). Normal CSF and neurophysiological testing do not exclude PCB in early disease stage (3,11). Therefore, we should not delay diagnosis and treatment if GBS or its variants are suspected on clinical grounds (9). MRI and CSF analysis were normal in our case. The strongest argument for PCB diagnosis is the presence of anti-GT1a-IgG antibodies (3,6,11). Some of which might cross-react with GQ1b (9). Patients with anti-GT1a antibodies develop PCB because a GT1a ganglioside, in addition to being abundantly detected at the cranial nerves such as the glossopharyngeal and vagus nerves (4), is also present in the oculomotor nerves (11). In 100 patients with PCB, Anti-GT1a IgG and Anti-GQ1b IgG antibodies were positive in 51.0% and 39.0% of the patients respectively (6). Increased titers of immunoglobulin M (IgM) antibodies to sulfatide have often been reported in patients with immune mediated neuropathies. But a selective reactivity to sulfatide, however, is rarely found and is associated with different forms of neuropathies, limiting its usefulness in the diagnosis of these latter (1). Determination of antiganglioside antibodies in our patient revealed positive Anti-sulfatide IgM antibodies but we did not know how to interpret this result because Anti-GT1a IgG and Anti-GQ1b IgG antibodies that we expected to find were negative. The general principle of GBS management includes the use of intravenous immunoglobulin or plasma exchange. In addition, nasogastric feeding and ventilator support can be useful if bulbar and respiratory functions are impaired (11). It has been reported that the ptosis as well as dysphagia associated with the PCB variant can be improved by administration of acetylcholine esterase inhibitor (4). In the largest series, patients with pure PCB were more likely to require intubation than those with overlap syndromes and this correlated with degree of bulbar involvement (11). Our patient was not intubated and his symptoms were improved after immunoglobulin therapy with complete recovery after 6 weeks. CONCLUSION The PCB syndrome is a regional axonal variant of GBS not known by most neurologists. Our case, whose clinical presentation was misleading initially, highlights the diagnosis difficulty of this entity. However, the recognition of this syndrome based on core clinical features (history of infection, monophasic disease course, and symmetrical cranial or limb weakness) is important to establish early correct diagnosis, avoid unnecessary investigations and guide appropriate use of immunotherapy (10). CONSENT FOR PUBLICATION: The patient gave a written consent for publication. AUTHOR ROLES: Hicham El Otmani and Vicky Fotso worked at the Conception and writing of the manuscript. Mohamed abdoh Rafai and Bouchra Moutawakil : have read and criticized the manuscript. All authors read and approved the final manuscript. COMPETING INTERESTS The authors declare no competing interest. 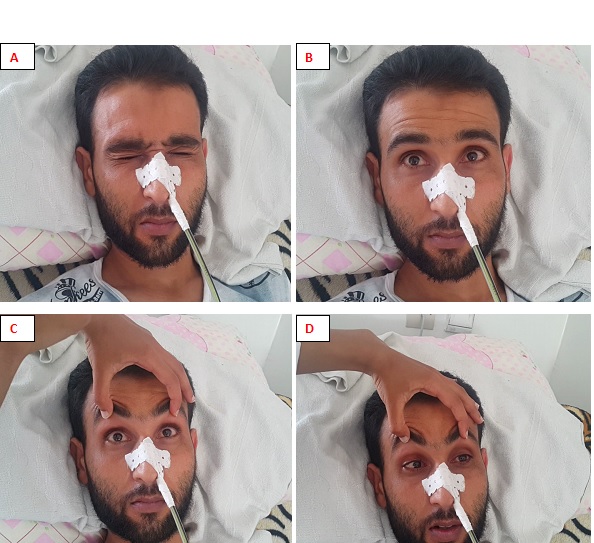 Fig 1: patient at admission: the oculomotor nerves III and VI as well as the VII nerve are impaired; A : mild bilateral facial palsy ; B: left internal strabismus (paresis of left VI nerve) C : gaze upward paralysis (bilateral III nerve) D : left eye abduction paresis.
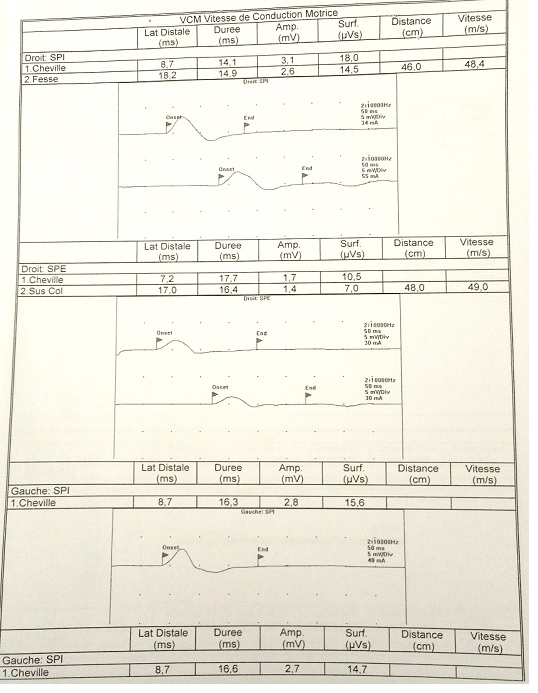 Fig 2: EMG showing an elongation of distal latencies with a decrease in the amplitude of motor potential in lower limbs.
 Fig 3: patient 6 weeks later: cranial nerves examination is normal; A: eyelid orbicularis muscle (VII nerve); B: right VI nerve and left III nerve; C: III nerve ; D: left VI nerve and right III nerve. letter PAANS congress Eldoret Kenya 2021 APPORT DES EXPLORATIONS RADIOLOGIQUES ET NEUROPHYSIOLOGIQUES DANS LE BILAN DES NEVRALGIES CERVICO-BRACHIALES OBSERVEES DANS UN LABORATOIRE DE NEUROPHYSIOLOGIE CLINIQUE A DAKAR AU SENEGALINTRODUCTION La Névralgie cervico-brachiale (NCB) se définit comme une douleur naissant au cou et irradiant le long du membre supérieur, secondaire à une irritation de la partie proximale d’un nerf spinal cervical. Les causes peuvent être multiples et/ou intriquées mais la littérature en retient trois principales: la hernie discale, l’ostéophytose postérieure et l’uncarthrose souvent associée à des lésions dégénératives discales et l’arthrose interapophysaire postérieure (1;4). L’imagerie occupe une place de choix pour le diagnostic étiologique de la NCB, mais elle peut parfois être normale. Même si toutes les radiculalgies ne s’accompagnent pas d’anomalies électrophysiologiques (16), l’Electro-Neuro-Myographie (ENMG) (3,9,18) et les Potentiels Evoqués Somesthésiques (PES) dans le cas où l’ENMG est en défaut (13), peuvent aider au diagnostic. En effet, ces examens neurophysiologiques peuvent être normaux si les fibres nerveuses atteintes sont de petite taille ou s’ils sont réalisés trop précocement, la dégénérescence des axones ou les lésions des gaines de myéline nécessitant plusieurs semaines (14). L’ENMG et les PES constituent le prolongement de l’examen clinique neurologique. Ils permettent de distinguer une lésion radiculaire d’une lésion plexique, de situer la lésion, d’en préciser la sévérité et surtout d’écarter une atteinte tronculaire distale isolée ou associée, pouvant mimer la symptomatologie d’une NCB (14,5). L’objectif de notre étude était de montrer l’apport des différents examens, radiologiques, ENMG et PES dans le diagnostic des NCB. METHODOLOGIE L’étude s’est déroulée dans un laboratoire privé de Neurophysiologie clinique à Dakar au Sénégal. Il s’est agi d’une étude rétrospective, sur une période d’une année, du 1er Janvier au 31 Décembre 2018. Elle a concerné les patients orientés par leur médecin traitant au laboratoire pour une exploration dans le cadre d’une suspicion clinique de NCB. Nous avons inclus dans l’étude tous les patients dont les résultats de l’imagerie cervicale étaient disponibles sur la demande d’ENMG couplé aux PES. Nous avons analysé les paramètres neurophysiologiques suivants : les latences et les amplitudes des potentiels moteurs et sensitifs, les vitesses de conduction nerveuse motrice et sensitive, la latence de l’onde F aux membres supérieurs et en fonction de l’atteinte clinique l’aspect du tracé obtenu à l’EMG de détection à l’aiguille. Les troncs nerveux systématiquement étudiés ont été les nerfs médian, ulnaire et radial. Les muscles les plus souvent testés à la myographie ont été le deltoïde, les sus et sous épineux, le biceps brachial, le triceps brachial, le court abducteur du pouce, le premier interosseux et l’extenseur propre de l’index. Les PES des membres supérieurs ont été recueillis par la technique de stimulation avec des bagues placées successivement sur les nerfs digitaux des patients, le pouce pour la racine C6, le majeur pour la racine C7 et l’auriculaire pour la racine C8. Cette technique qui contrairement à la stimulation des nerfs mixtes, permet de stimuler sélectivement les fibres sensitives, a permis d’analyser les ondes N9 (réponse plexuelle), N13 (réponse de jonction cervico-bulbaire) et N20 (réponse corticale). RESULTATS Dix-neuf patients ont pu bénéficier d’un ENMG couplé aux PES. La plainte fonctionnelle en rapport avec la NCB, pouvait être unique ou multiple chez le même patient. Parmi ces 19 patients, seuls 14 avaient une imagerie cervicale et donc répondaient entièrement à nos critères d’inclusion. Ils se plaignaient majoritairement de paresthésies à type de décharge électrique le long du membre supérieur concerné (10 patients) et de douleurs cervicales ou scapulaires (04 patients). L’ENMG de stimulo détection et de détection était normal pour 13/14 patients. Dans 1 cas il était anormal et objectivait une atteinte tronculaire sensitivo motrice axonale du nerf ulnaire objectivée par la diminution de l’amplitude du potentiel sensitif et du potentiel moteur comparativement à la neurographie controlatérale du même nerf. Les résultats des PES sont représentés par la figure 1. Ils étaient normaux pour un patient qui avait aussi l’ENMG normal et la radiographie cervicale normale. Pour les 13 autres patients chez qui les PES étaient altérés, ils mettaient en évidence une atteinte poly-radiculaire caractérisée par une latence normale de l’onde N9, associée à un allongement de la latence de l’onde N13 (13 patients/14) ou une plexopathie caractérisée par un allongement de la latence de l’onde N9 avec répercussion sur la latence de N13 (01 patient/14). La figure 2 est une iconographie de PES d’un patient avec polyradiculopathie C6-C8. Le patient avec l’ENMG en faveur de l’atteinte tronculaire du nerf cubital avait en plus une radiculopathie C6-C8 mise en évidence par les PES. Le bilan radiologique des 14 patients, comportait une IRM cervicale (09 patients), une TDM cervicale avec ou sans injection de gadolinium (02 patients) et une radiographie standard du rachis cervical (03 patients). Les 03 patients avec radiographie normale avaient également un ENMG normal, des PES normaux dans 1 cas et des PES en faveur d’une polyradiculopathie dans 2 cas. Les 2 TDM cervicales étaient anormales avec des anomalies pouvant être multiples chez un même patient : scoliose, arthrose étagée et ostéophytes étagés. Chez ces 2 patients, l’ENMG était normal mais les PES montraient un tableau de polyradiculopathie. Sur les 9 IRM cervicales, 3 étaient normales et 6 étaient anormales avec des lésions très souvent variées chez le même patient et dominées par la saillie discale cervicale étagée (04 cas), suivie de la cervico-uncarthrose étagée (03 cas) et plus rarement de la sténose de trou de conjugaison (01 cas), d’une protrusion discale postéro-médiane (01cas) et d’un canal cervical étroit (01 cas). Les PES étaient anormaux chez tous les 9 patients ayant eu une IRM cervicale, en faveur d’une polyradiculopathie, même lorsque l’IRM était normale (3 patients sur 9). DISCUSSION Le faible nombre de patients de l’étude s’explique par le biais de sélection de la population d’étude. En effet, l’exploration neurophysiologique ne fait pas partie de façon courante du bilan d’exploration des NCB, la majorité des patients étant orientée vers l’imagerie d’emblée. Le manque de certains détails concernant les signes cliniques ou les incidences des examens radiographiques est lié au fait que ces notions ont été relevées à partir des éléments rapportés sur le bulletin d’examen par le médecin traitant. L’ENMG, examen le plus fréquemment réalisé devant une NCB, était normal pour la quasi-totalité des patients (13/14 patients). Ce résultat peut s’expliquer par le mécanisme à la base de la NCB et le délai de réalisation de l’ENMG par rapport au début de la symptomatologie. En effet, selon certains auteurs, lors de la section d’un nerf périphérique, sensible, moteur ou mixte, les fibres nerveuses du bout central resteraient normales au point où des signes de dégénérescence liés au traumatisme ne se verraient pas avant plus d’un mois et demi à 2 mois (14). Par ailleurs la topographie de l’atteinte et la nature des fibres lésées expliquent le résultat souvent normal de l’ENMG, les lésions radiculaires qui sont proximales au ganglion rachidien préservent leur potentiel sensitif en périphérie alors que dans les lésions plexiques et tronculaires qui sont distales par rapport au ganglion rachidien, les potentiels sensitifs sont altérés. L’ENMG de détection est performant dans le diagnostic des NCB, à condition qu’il y ait une souffrance axonale même infraclinique de la racine motrice. En outre, en cas de symptômes uniquement sensitifs, l’ENMG peut être strictement normal (3). Le réflexe segmentaire H, dont l’intérêt est beaucoup plus connu dans la neurographie aux membres inférieurs, n’a pas été exploré dans notre étude. Il aurait pu apporter des arguments en faveur de la NCB. En effet, certains travaux sur les radiculopathies du membre supérieur, lui accordent une sensibilité et une spécificité similaire à celle de la neuro imagerie (12). Dans notre étude, les PES ont apporté le plus d’arguments en faveur de la NCB (13/14 patients), alors que leur utilité dans le diagnostic de la radiculopathie reste controversée. Dans une étude portant sur l’apport de l’électrophysiologie dans la cervicalgie, les auteurs rapportent que de façon assez exceptionnelle, dans le cadre d’une radiculopathie, il pouvait arriver que les PES soient anormaux alors que l’exploration ENMG était normale (17). Le déficit moteur serait le mieux corrélé aux ENMG anormaux, tandis que les PES seraient anormaux le plus souvent lorsque le déficit sensitif prédominait (8). Les PES par la stimulation nerveuse cutanée constitueraient donc un complément utile aux méthodes électrophysiologiques conventionnelles d’évaluation des radiculopathies, en particulier en l’absence de déficit moteur. La technique de stimulation des nerfs digitaux que nous avons utilisée, explorant de manière plus spécifique le versant sensitif, pourrait expliquer ce meilleur rendement diagnostic. Pour Boulu, les PES des nerfs cutanés et dermatomaux seraient plus spécifiques de l’atteinte d’une racine, même s’ils sont beaucoup plus difficiles à obtenir que les PES de nerfs mixtes, pluriradiculaires, qui sont souvent normaux lorsqu’une seule racine est lésée (3). Le progrès réalisé dans le domaine de l’imagerie a augmenté le choix des techniques d’exploration. Cependant toutes ont leur intérêt mais aussi leur limite. Ainsi, la radiographie standard reconnue comme étant moins sensible que les autres moyens d’imagerie, peut apporter des arguments en faveur de NCB. Les incidences de radiographies, face, profil et ¾ permettent de confirmer la présence de lésions dégénératives, de vérifier la concordance du niveau lésionnel clinique lorsqu’il est typique et de rechercher d’autres causes éventuelles intriquées (1,15,6). Dans notre étude, cet examen, n’a pas permis d’objectiver des anomalies en faveur de la NCB. Cela pourrait être dû au fait que la radiographie visualise mal les lésions des parties molles, que certaines incidences n’aient pas été réalisées et que l’interprétation des clichés peut ne pas être reproductible d’un radiologue à l’autre. Par ailleurs la présence des lésions de discopathie dégénérative à l’étage cervical est très fréquente chez le sujet asymptomatique de telle sorte qu’il est difficile d’affirmer leur caractère pathogène (1). En présence d’anomalies électrophysiologiques de type plexique couplées à la symptomatologie clinique de NCB le bilan devrait être complété par une imagerie du thorax à la recherche d’une pathologie des apex pulmonaires notamment tumorale, d’une côte surnuméraire ou d’une apophysomégalie transverse C7 dans le cadre du diagnostic différentiel d’un syndrome du défilé cervico-thoraco-brachial (7). Tous les patients ayant réalisé une TDM cervicale, avec et sans injection de Gadolinium, présentaient des anomalies à l’imagerie pouvant expliquer la NCB. La scanographie cervicale sans injection de produit de contraste permettrait une bonne individualisation des rétrécissements dégénératifs osseux canalaires et foraminaux ; Par contre, l’individualisation de l’espace épidural et du couple racine-veine est mieux appréhendée par une scanographie avec injection de produit de contraste intraveineux (1). La TDM avec injection de produit de contraste est un examen performant pour visualiser une compression radiculaire d’origine discale ou dégénérative et pour apprécier les dimensions du canal rachidien. Dans notre série l’IRM cervicale, l’examen le plus réalisé (9/14 patients) était anormal chez 06 patients. Il a permis d’objectiver des anomalies à type majoritairement de hernie discale (4/6 patients) et de cervico-uncarthrose (03 patients) ce qui corrobore les données de la littérature (1,15,11). L’IRM individualise très bien morphologiquement la moelle spinale mais aussi les anomalies de signal, les disques (pincement, hernie, changement de signal). La sensibilité de l’IRM est considérée comme importante pour diagnostiquer les discopathies dégénératives et visualiser les ligaments jaunes (1). Concernant l’imagerie dans la NCB, un consortium de recommandations de pratique clinique, stipule que l’IRM serait l’examen le plus performant. La scanographie avec injection permettrait une bonne analyse du disque et des ostéophytes mais elle est contre-indiquée chez les patients intolérants aux produits de contraste iodés et pourrait être ininterprétable au niveau des espaces C7-T1 voire C6-C7, chez les patients trapus et/ou ayant un cou court à cause des artéfacts. L’IRM ne présenterait pas les mêmes contre-indications allergiques et est peu gênée par la morphologie des patients, mais elle est responsable de faux négatifs concernant l’analyse des ostéophytes et des hernies foraminales (1). La combinaison de l’IRM cervicale à la radiographie standard reste la meilleure alternative notamment pour le bilan pré chirurgical (10). L’exploration neurophysiologique a permis de confirmer la radiculopathie chez 5 patients sur 6 qui avaient une imagerie normale. Dans les NCB, les hernies cervicales affectent surtout les disques cervicaux les plus mobiles, à savoir C5-C6 et C6-C7 (15). Cela explique que les sixième, septième voire huitième racines soient les plus souvent concernées comme le démontrent les anomalies des PES retrouvées chez ces patients avec imagerie normale, faites essentiellement de radiculopathies C6, C7 et C8. L’imagerie, en particulier l’IRM, montre des images précises des lésions grâce à sa bonne résolution spatiale, les techniques électrophysiologiques ajoutent une résolution temporelle qui permet de mieux apprécier le retentissement neurologique de lésions passées inaperçues à l’imagerie (2). Cependant, tous ces différents examens peuvent rester normaux bien que la plainte fonctionnelle de la NCB soit présente, comme en témoigne le cas d’un patient dans notre étude dont tout le bilan paraclinique (explorations neurophysiologiques et radiologiques) n’a pas objectivé d’anomalies. CONCLUSION Le diagnostic positif de NCB est avant tout clinique. Les examens complémentaires radiographiques ou neurophysiologiques, apportent une aide pour confirmer le diagnostic, faire le diagnostic différentiel et/ou étiologique surtout lorsqu’ils sont couplés. Ils ont un intérêt particulier si un geste chirurgical est envisagé. Les PES sont utiles pour authentifier l’organicité d’un symptôme subjectif notamment sensitif particulièrement lorsque l’ENMG est normal. Mais il demeure important de garder en tête que la négativité des examens neurophysiologiques n’exclut pas le diagnostic de NCB.  Figure 1 : Résultats des PES
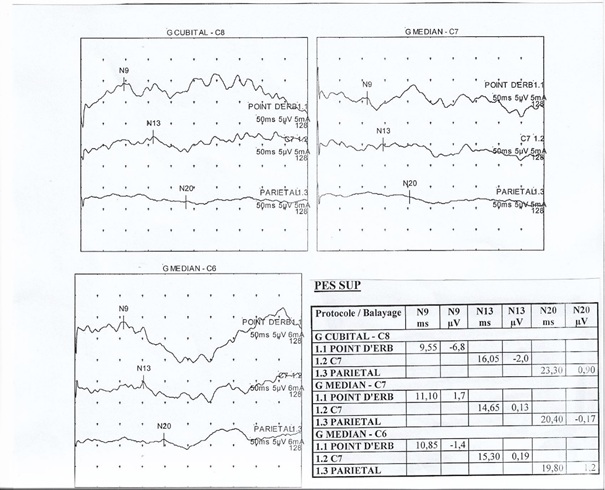 Figure 2 : Iconographie d’une radiculopathie C6-C8 aux PES INTRODUCTION L’épilepsie est un dysfonctionnement cérébral caractérisé par une prédisposition chronique à la génération de crises épileptiques et par les conséquences neurobiologiques, cognitives, psychologiques et sociales de cet état. La définition de l’épilepsie nécessite la survenue d’au moins une crise épileptique. Une crise épileptique est la survenue transitoire de signes et symptômes liés à une activité neuronale anormale, excessive ou synchrone, dans le cerveau. Un syndrome épileptique définit une forme d’épilepsie caractéristique par l’âge de survenue des crises, son évolution dans le temps, le type de crise et surtout par le motif électroencéphalographique. Les données de l’électroencéphalogramme (EEG) sont capitales dans cette classification. Le syndrome de West ou encéphalopathie infantile avec hypsarythmie est une forme rare d’épilepsie. Il débute le plus souvent avant un an chez un nourrisson avec un retard de développement psychomoteur préalable ou chez un nourrisson normal avec un arrêt du développement psychomoteur, puis régression [16]. Le pronostic est étroitement lié à la pathologie sous-jacente et à la précocité du traitement [5]. Au Sénégal, le syndrome de West demeure l’encéphalopathie épileptique la plus fréquente du nourrisson. Il est principalement secondaire à des causes périnatales, favorisées par un mauvais suivi prénatal .C’est ainsi que nous avons décidé de réaliser cette étude dont l’objectif principal était de décrire les caractéristiques du syndrome de West. PATIENTS ET METHODES Il s’est agi d’une étude rétrospective et descriptive, qui s’est déroulée à la clinique neurologique du Centre Hospitalier Universitaire de Fann, et à l’Hôpital national pédiatrique Albert Royer, à Dakar-Sénégal. Nous avons inclus les enfants suivis pour un syndrome de West entre Avril 2012 et Aout 2018. Les enfants souffrant d’une encéphalopathie épileptique sans hypsarythmie et ceux ayant des dossiers incomplets, étaient exclus de l’étude. A l’aide de dossiers médicaux, nous avons recueilli les données biographiques (âge, sexe), les données cliniques relatives au syndrome de West, les données électro-encéphalographiques, les données radiologiques (Tomodensitométrie cérébrale, Imagerie par résonnance magnétique), les données thérapeutiques et évolutives. Les différents paramètres étudiés étaient : l’âge au moment du diagnostic, l’âge d’apparition des spasmes, la nature des spasmes, les autres types de crises épileptiques, le développement psychomoteur, le pattern électro-encéphalographique, les modalités thérapeutiques et évolutives. Les données ont été saisies sur Microsoft Word. Nous avons procédé aux calculs de fréquence, de la moyenne, de la médiane, de l’écart-type. L’analyse des données a été faite sur Microsoft Excel 2010. RESULTATS Nous avons colligé dans cette étude 37 nourrissons, dont 15 filles (41%) et 22 garçons (59%), avec un sex ratio à 1,47. Les âges extrêmes étaient de 3 et 24 mois. La moyenne d’âge était de 8,8 mois ± 4,04 mois, avec une médiane de huit mois. L’âge moyen d’apparition des spasmes était de 3,1 mois avec des extrêmes de 2 et 7 mois. Le suivi prénatal avait été réalisé chez une seule femme en gestation. La grossesse a été menée à terme chez 15 enfants (40,54%), et un enfant était issu d’une grossesse prématurée (2,70%). Dix-sept enfants (45,95%) étaient nés d’un accouchement eutocique, et six (16,21%) d’un accouchement dystocique. Nous avons noté des antécédents de souffrance néonatale chez sept nourrissons (18,91%). Un nourrisson (2,70%) avait des antécédents de trisomie 21. Les spasmes épileptiques étaient présents chez tous les nourrissons. Ils apparaissaient entre un et sept mois de vie. Ces spasmes étaient en flexion chez 26 enfants (72,97%), en flexion et extension chez deux nourrissons (5,40%). Leurs caractéristiques n’étaient pas précisées chez les autres nourrissons. Les autres types de crises sont représentés dans le tableau I. Un patient avait présenté des convulsions néonatales avant le début des spasmes. Le développement psychomoteur était altéré chez 29 patients (78,37%). Le contact social était pauvre chez 17 nourrissons (45,95%). Il était bon chez quatre enfants (10,81%). La microcéphalie était retrouvée chez deux nourrissons (5,40%). Nous avons représenté les autres signes neurologiques associés dans le tableau II. L’électroencéphalogramme était réalisé chez tous les patients, et montrait une hypsarythmie (figures 1 et 2a) chez tous les nourrissons (100%). Les signes radiologiques étaient étudiés au scanner cérébral (tableau III), et/ou à l’Imagerie par résonnance magnétique qui était normale chez trois nourrissons (8,11%). Elle montrait une hydrocéphalie chez un nourrisson (2,70%) et une encéphalomalacie post-anoxique chez un nourrisson (2,70%). Les molécules utilisées pour le traitement étaient : le phénobarbital, le valproate de sodium, la carbamazépine, le clonazépam, le diazépam, le piracétam, la bétaméthasone et la prednisone. La corticothérapie était de courte durée associée au traitement adjuvant à base de calcium. En première intention, (Tableau IV), les nourrissons étaient traités à base d’une monothérapie, une bithérapie, ou une trithérapie. La kinésithérapie était associée au traitement médicamenteux. L’évolution clinique sous traitement était variable (figure 3). Les acquisitions psychomotrices s’étaient améliorées chez quatre enfants (10,81%). L’électroencéphalogramme était devenu quasi normal (figure 2b) chez deux nourrissons (5,40%). DISCUSSION Notre étude révélait une prédominance masculine. Nos données sur la prédominance masculine, l’âge au moment du diagnostic (3 à 24 mois) et l’âge d’apparition des spasmes (2 à 7 mois) sont globalement comparables à ceux retrouvés dans la littérature. Au Canada, en 2001, Brna et al. avaient noté une prédominance masculine à 60%. En 2017, au Kosovo, Zeka et al. [17] avaient également retrouvé une prédominance masculine, avec une moyenne d’âge d’apparition des spasmes de 2,5 mois. Au Portugal, Matta et al. [13], en 2007, avaient noté une prédominance masculine à 62%, avec une moyenne d’âge au moment du diagnostic de 4,9 mois. En Tunisie, Dhahri et al. [5] avaient retrouvé une moyenne d’âge au moment du diagnostic de 8,1 mois. En Afrique du Sud, Keshave et al. [12] en 2017, avaient aussi obtenu une prédominance masculine (87,5%) sur un échantillon de huit patients. Ils rapportaient un âge moyen de début des spasmes de 4,4 mois avec des extrêmes de 1 et 9 mois. Et la moyenne d’âge au moment du diagnostic était de 7,5 mois avec des extrêmes de 3 et 18 mois. Une étude réalisée précédemment au Sénégal en 2017, par Halima et al. [10] montrait une moyenne d’âge d’apparition des spasmes de 6 mois, avec un âge moyen de 10 mois au moment du diagnostic. Le taux de suivi prénatal était bas, d’après nos résultats. En effet, il n’avait été noté que dans un seul cas (2,70%). La consultation prénatale permet de prendre les mesures appropriées pour que l’accouchement se déroule au bon moment (programmer si nécessaire), au bon endroit (référer s’il le faut), et dans les meilleures conditions (considérer les particularités de chaque parturiente) [6]. Les consultations prénatales au troisième trimestre seraient l’occasion de faire un pronostic de l’accouchement grâce à l’utilisation du score de risque de dystocie [14]. Le taux de naissances par accouchement eutocique était élevé. Ce taux de naissance élevé par accouchement eutocique était aussi observé en Afrique du sud par Keshave et al. [12]. Les facteurs de risque retrouvés dans notre série étaient par ordre croissant, l’absence de suivi prénatal, la souffrance néonatale, l’accouchement dystocique et la prématurité. Dans notre étude, tous les patients présentaient des spasmes avec une prédominance des spasmes en flexion. Le syndrome de West est une encéphalopathie épileptique qui se traduit par des spasmes en flexion plus fréquents que les spasmes en extension et les spasmes mixtes. Les spasmes mixtes associent la flexion des membres supérieurs et l’extension des membres inférieurs [16]. Les spasmes en flexion ont la même valeur sémiologique que les spasmes en extension. Plus de la moitié des patients avaient un développement psychomoteur altéré. Le contact social était pauvre dans la moitié de notre échantillon. Keshave et al. [12] avaient observé des troubles du développement psychomoteur chez tous les enfants (100%). Zeka et al. [17] avaient aussi noté des troubles du développement psychomoteur dans 13 cas (78,6%). L’électroencéphalogramme était anormal, comportant une hypsarythmie chez tous les nourrissons. Keshave et al. [12], avaient également observé une hypsarythmie sur l’électroencéphalogramme chez tous les patients. Dhahri et al. [5], notaient une hypsarythmie typique sur l’électroencéphalogramme chez 21 enfants dans un échantillon de 30 cas. Dans notre série, les causes étaient aussi variées, avec une prédominance de l’atrophie cortico-sous-corticale (21,62%), suivie de l’atrophie fronto-pariétale (10,81%), des séquelles d’ischémie cérébrale (10,81%) et des malformations cérébrales. Les autres causes les moins fréquentes étaient l’hémorragie cérébrale, l’encéphalomalacie post-anoxique. L’analyse génétique à la recherche d’aberration chromosomique n’a pas été réalisée dans notre étude. Dans la série de Keshave et al. [12], les anomalies radiologiques étaient dominées par l’atrophie cortico-corticale et l’agénésie du corps calleux. Dhahri et al. [5] avaient noté des examens neuroradiologiques pathologiques chez 22 cas et deux enfants avaient une aberration chromosomique, sur un échantillon de 30 cas. Selon la littérature [4], les étiologies du syndrome de West sont très variées, identifiées dans plus de 50% des cas. Les causes les plus fréquentes, par ordre décroissant, sont l’encéphalopathie hypoxique-ischémique (10%), les anomalies chromosomiques (8%), les malformations (8%), l’accident vasculaire cérébral périnatal (8%), la sclérose tubéreuse de Bourneville (7%) et la leucomalacie périventriculaire, ou l’hémorragie cérébrale (5%). En première intention [tableau IV], les nourrissons recevaient majoritairement une bithérapie à base de valproate de sodium et de corticoïdes (bétaméthasone ou prednisone). En monothérapie, la molécule la plus prescrite était le valproate de sodium, suivie du phénobarbital. La pharmacorésitance nécessitait de faire recours en deuxième intention [tableau V], majoritairement à une bithérapie à base de valproate de sodium et corticoïdes, ou valproate de sodium et vigabatrin. Les corticoïdes administrés étaient la prednisone ou la bétaméthasone. Dans l’échantillon de Keshave et al. [12], tous les huit enfants (100%) avaient reçu du valproate de sodium. La moitié des enfants avait reçu une corticothérapie orale à base de prednisone. Et l’hormone adrénocorticotrophique était administrée chez 3 enfants. Seuls deux enfants avaient reçu du vigabatrin. Dans la série d’Ayadi et al. [1], les patients recevaient une association de plus de deux antiépileptiques en cas de persistance des crises. Dans l’étude de Dhahri et al. [5], le traitement était basé essentiellement sur le vigabatrin et les corticoïdes. Des études récentes suggèrent que l’initiation précoce du traitement, soit avec un traitement hormonal ou le vigabatrin, améliore à long terme les troubles cognitifs. En outre, il a été observé une meilleure efficacité de l’ACTH comparé au vigabatrin dans les troubles cognitifs pour les formes cryptogéniques. Différents corticostéroïdes (hydrocortisone, prednisone, prednisolone ou dexaméthasone), et antiépileptiques peuvent être utilisés en deuxième intention lorsque les médicaments de première ligne sont inefficaces ou contre-indiqués. Environ 25 à 40% des patients dont les spasmes persistent, avec un retard psychomoteur, peuvent être candidats à la chirurgie [4]. La chirurgie n’est pas indiquée si la résection corticale peut créer un nouveau déficit neurologique, en cas de lésions cérébrales diffuses ou de maladie métabolique [9]. Dans notre série, l’évolution clinique [figure 3] était surtout marquée par un arrêt des crises (24,32%) et une persistance des crises chez 18,92%. Et au cours de la surveillance paraclinique, seuls deux de nos patients (5,40%) avaient un électroencéphalogramme de suivi normal. Keshave et al. [12] avaient noté un arrêt des crises dans la moitié de leur échantillon. La réduction de la fréquence des spasmes était aussi retrouvée chez la moitié des patients. Le devenir du syndrome de West est difficilement prévisible, cette encéphalopathie étant un grave syndrome souvent réfractaire au traitement et fréquemment associé à un retard mental [7]. CONCLUSION Le syndrome de West est une encéphalopathie épileptique fréquente chez le nourrisson. Le tableau clinique est dominé par des spasmes en flexion. Les crises associées sont majoritairement représentées par les myoclonies. Les facteurs de risque sont dominés par l’absence de suivi prénatal, la souffrance néonatale, l’accouchement dystocique. La polythérapie permet d’arrêter les crises, mais certains cas peuvent demeurer pharmacorésistants. Dans notre contexte, l’imagerie par résonnance magnétique n’est pas disponible chez la majorité des enfants, de même que le bilan génétique. Le vigabatrin est rarement prescrit à cause de son inaccessibilité et de son coût élevé dans nos régions. 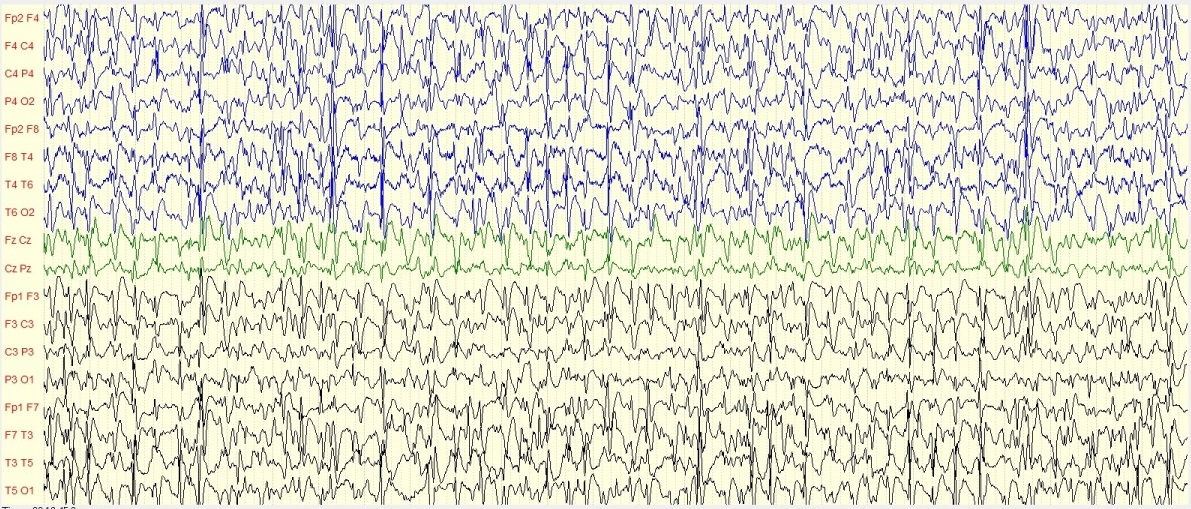 Figure 1 : Tracé EEG de sommeil montrant une hypsarythmie avec des pointes-ondes généralisées chez une fillette de 24 mois
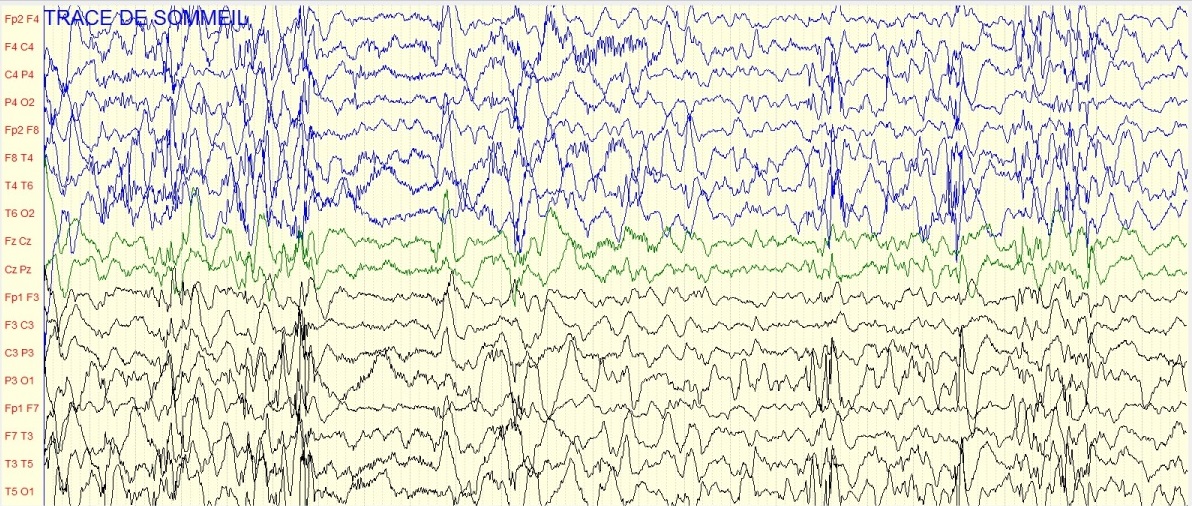 Figure 2a : Tracé EEG de sommeil montrant une hypsarythmie chez un garçon de 11 mois
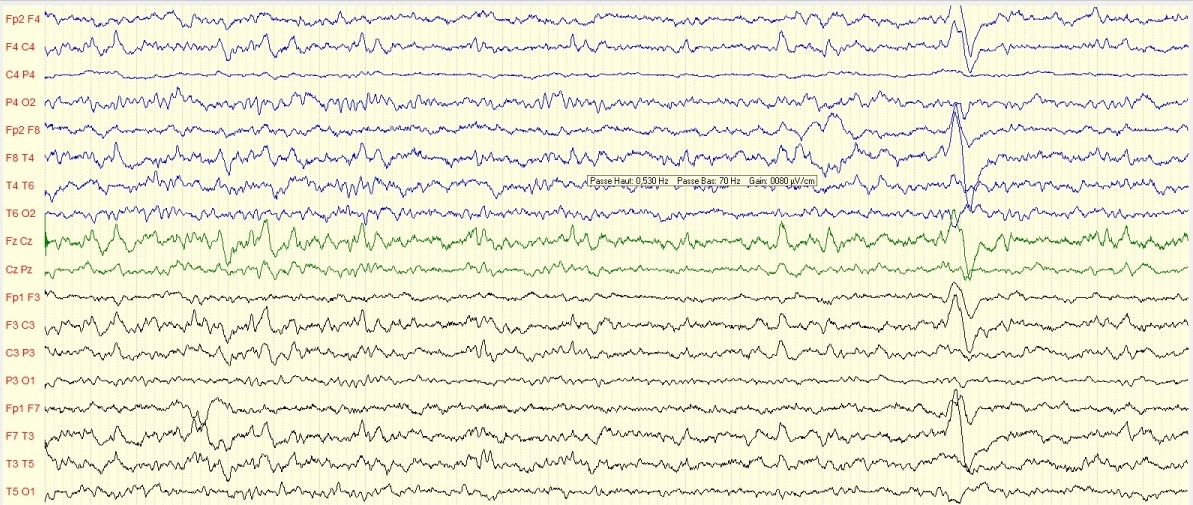 Figure2b : Tracé EEG de contrôle quasi-normal avec disparition de l’hypsarythmie et des ébauches de figures physiologiques du sommeil
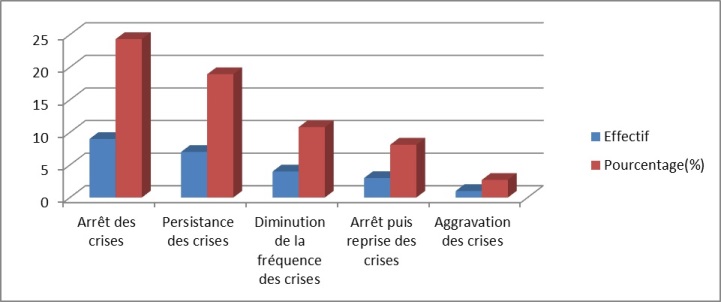 Figure 3 : Aspects évolutifs Tableau I : Autres types de crises
Tableau II : Signes neurologiques associés
Tableau III : Signes radiologiques
Tableau IV : Schéma thérapeutique en première intention
Tableau V : Schéma thérapeutique en deuxième intention
Articles récents
Commentaires récents
Archives
CatégoriesMéta |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
© 2002-2018 African Journal of Neurological Sciences.
All rights reserved. Terms of use.
Tous droits réservés. Termes d'Utilisation.
ISSN: 1992-2647