
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
INTRODUCTION L’accident vasculaire cérébral (AVC) est moins fréquent chez les enfants comparés aux adultes (12). Plusieurs travaux ont été faits dans les pays en voie de développement (8,11) cependant les publications en Afrique demeurent rares (22,21,20,19,26). Au Sénégal quelques études sur les AVC (20,19,26) de l’enfant ont été menées. L’objectif de notre travail était de déterminer l’épidémiologie, les aspects cliniques, paracliniques et les facteurs de risque des accidents vasculaires cérébraux de l’enfant à Dakar (Sénégal). PATIENTS ET METHODE Nous avons mené une étude multicentrique, rétrospective et descriptive dans les services de pédiatrie de l’Hôpital d’enfants Albert Royer et de Diamniadio et au Service de Neurologie du Centre hospitalier national universitaire Fann. Elle portait sur les dossiers des enfants hospitalisés pour accident vasculaire cérébral entre Janvier 2005 et Janvier 2020. Ont été inclus tous les dossiers des enfants âgés de 2 mois à 18 ans chez qui le diagnostic clinico-radiologique d’accident vasculaire cérébral a été retenu. Les dossiers avec des informations incomplètes ont été exclus. Les variables étudiées étaient :
Tous les patients étaient suivis par la même équipe médicale faite de neuropédiatres, pédiatres et radiologues. L’analyse des données recueillies a été réalisée avec le logiciel Excel 2016. Des analyses univariées ont été effectuées pour le calcul des fréquences et des moyennes. RESULTATS Deux cent quarante (240) dossiers d’accident vasculaire cérébral ont été colligés sur une période de 15 ans, 201 cas d’infarctus cérébral (83,75%) et 39 cas d’hémorragie cérébrale (16,25%). Ils étaient 140 garçons (58,33%) et 100 filles (41,66%) soit un sex-ratio de 1,4. Cette prédominance masculine était retrouvée dans les infarctus cérébraux (58,21%) et les hémorragies cérébrales (61,54%) (Figure 1). Tous les âges pédiatriques étaient représentés : nourrissons (20%), enfants (56,25%) et adolescents (23,75%). Les enfants sont majoritaires aussi bien dans le groupe des AVCI (60,2%) que dans celui des AVCH (35,9%) (Tableau 1). L’âge moyen de survenue de l’AVC était de 60 mois (extrêmes 3 mois et 204 mois), de 67 mois dans les AVCI et de 46 mois dans les AVCH. Le mode de survenue de l’AVC était brutal dans la majorité des cas (94,5%) ou rapidement progressif (5,5%). Les principales manifestations cliniques étaient l’hémiplégie (76,3%), les convulsions (20,4%) et les troubles de la conscience (5%) (Tableau 2). Trois patients avaient des signes extra-neurologiques : deux patients avaient une insuffisance cardiaque droite avec hépatomégalie et reflux hépato-jugulaire, un autre avait des œdèmes de type rénal. L’imagerie cérébrale (Scanner cérébral et/ou Imagerie par résonnance magnétique) était réalisée chez tous nos patients. Dans les infarctus cérébraux l’artère cérébrale moyenne était la plus touchée (63,7%) suivie des artères cérébrales antérieure (13,4%) et postérieure (11%) (Tableau 3). L’atteinte de l’Artère cérébrale moyenne était unilatérale chez 113 patients et bilatérale chez 15 autres enfants. L’atteinte de l’artère cérébrale antérieure était unilatérale chez 24 patients et bilatérale chez 3 autres patients. Les ischémies de l’artère cérébrale postérieure étaient unilatérales dans 18 cas et bilatérales dans 4 cas. L’ischémie intéressait plus rarement d’autres artères cérébrales avec parfois une occlusion conjointe de 2 artères (Tableau 3). L’hémorragie était sus tentorielle (94,8%), plus rarement sous tentorielle (5,2%). L’hémorragie sus tentorielle était lobaire (48,7%) ou profonde (46,2%) (Tableau 4). L’hémorragie était intra-parenchymateuse pure chez 32 enfants (82,1%) et était associée à une hémorragie sous arachnoïdienne chez 7 enfants (18%). Les malformations vasculaires (38,5%) constituaient la première étiologie des AVCH suivies de l’hypertension artérielle (2 patients âgés de 12 ans et 14 ans respectivement), des hémopathies (1 cas de thrombopénie et 1 cas de leucémie myéloïde chronique) et d’1 cas d’endocardite infectieuse (Tableau 5). Les étiologies identifiées dans les infarctus cérébraux étaient la drépanocytose SS (41,3%), les cardiopathies (11%), les méningo-encéphalites (9%) et l’anémie (7%) (Tableau 6). L’âge moyen de survenue de l’AVCI chez les drépanocytaires SS était de 5,3 ans (extrêmes 20 mois et 15 ans). Tous les patients drépanocytaires SS avaient une anémie normochrome normocytaire (89,8%) ou hypochrome microcytaire (10,2). La drépanocytose a été révélée par l’AVCI chez 54 patients (61,4%) alors que 34 patients (38,6%) étaient connus et suivis pour drépanocytose au moment de la survenue de l’AVC. Chez 22 enfants drépanocytaires SS, des ischémies multiples d’âges différents ont été notées. Les cardiopathies emboligènes (Tableau 7) étaient congénitales (10 cas) ou acquises (12 cas) et représentaient la deuxième cause d’AVCI de l’enfant. L’âge moyen de survenue de l’AVCI était de 72 mois (extrêmes de 23 mois et 168 mois). Deux patients avaient des signes d’insuffisance cardiaque droite avec un reflux hépato-jugulaire et une hépatomégalie. Dix-huit cas de méningoencéphalites ont été diagnostiqués et représentaient la troisième cause d’AVCI de l’enfant. Il s’agissait de 2 cas de tuberculose neuro-méningée dont un sur terrain de VIH1, un cas de méningite à Cryptococcus neoformans sur terrain de VIH1, un cas de pneumocoque et 14 cas de méningoencéphalite d’étiologie indéterminée dont 7 cas sur terrain de VIH1. L’anémie hypochrome microcytaire (13 cas) ou normochrome normocytaire (1 cas) était relativement fréquente. DISCUSSION : L’incidence des AVC chez l’enfant au Sénégal reste méconnue en dépit d’études antérieures (20,19,26). Dans la littérature, les hémorragies cérébrales sont rares chez l’enfant avec une incidence annuelle de 1,1 à 2,3/100000 habitants et par an comparé aux AVCI dont l’incidence annuelle est estimée entre 1,3 et 13/100000 habitants et par an (3,8). L’âge moyen dans les AVC dans notre étude était de 5 ans ce qui correspond aux données de la littérature où l’âge moyen varie entre 4 ans et 13 ans (7,18,4,16). Contrairement aux AVCI où la moyenne d’âge était de 67mois (M Ndiaye en 2018 retrouvait 71,5 mois) la moyenne d’âge est plus petite (3,83 ans) dans l’AVCH et reste inférieure aux données de la littérature où l’âge moyen des AVCH était de 5,25 ans avec des extrêmes entre 5,6 et 8,7 ans (26,15,10). La prédominance masculine est décrite par plusieurs auteurs (8,18,6) et dans notre étude la prédominance masculine variait entre 58,20% et 61,53% des cas selon le type d’AVC. La prédominance féminine est plus rare (21 ,16). Les manifestations cliniques dans les AVCH étaient dominées par l’hémiplégie (33,33%), hypertension intracrânienne 20,51%, les céphalées isolées 17,94% et les troubles de la conscience 15,38%. Nos résultats sont similaires aux données de la littérature où l’hypertension intracrânienne est rapportée entre 58 et 76% des patients et l’hémiplégie entre 16,2 et 62% des patients (8,10,17,23,24). La TDM cérébrale a permis de poser le diagnostic d’AVCH et 94,8% des lésions étaient sustentorielles ce qui était supérieur aux données de la littérature (73,4 et 92,85%) (8,13,28). Tout comme les hémorragies cérébrales, les hémiplégies (79,60%) constituent la principale manifestation clinique des infarctus cérébraux suivie des crises convulsives 18,05%, une aphasie 10,01% et des troubles de la conscience dans 2,4% des patients. Ndiaye et al en 2018 (19) avaient obtenu une nette prédominance de l’hémiplégie soit 84% suivi de l’aphasie 19% et des crises convulsives focales 10%. Les déficits neurologiques focaux sont aussi largement rapportés dans la littérature (16,1). L’ACM était la plus touchée (69,65%), suivie de l’ACA (30,34%) et l’ACP (15,92%). Ces résultats sont similaires aux données de la littérature avec une nette prédominance de l’ACM (8,20,19). La TDM cérébrale comme imagerie la plus accessible était demandée en première intention chez tous les malades et permettait de poser le plus souvent le diagnostic, l’IRM cérébrale était réalisée chez 11 patients. Dans notre contexte de sous-développement et d’inaccessibilité des explorations, nous rapportons 33,75% d’étiologie indéterminée chez les enfants victimes d’AVC dont 31,84% chez les enfants victimes d’AVCI ce qui est supérieur aux données de la littérature (9% à 22%) (12,13,23). La drépanocytose SS est la cause d’AVCI de l’enfant la plus représentée dans notre étude avec 41,29% des cas (20,19), c’est un problème de santé publique en Afrique subsaharienne (22,9) ; les données transversales en clinique ont rapporté une prévalence d’AVC de 2,9% à 16,9% chez les enfants atteints de drépanocytose (14). Notons que la drépanocytose est rarement rapportée comme étiologie d’AVCI dans les pays autres que l’Afrique avec une fréquence de moins de 11% dans les différentes études (3,9,25). Les cardiopathies emboligènes représentent moins de 7,7 à 33% des étiologies d’AVCI (20,19,16), certains auteurs rapportent une prédominance des cardiopathies congénitales (2,27) contrairement à notre étude où les cardiopathies acquises sont les plus représentées (12 cas) avec 4 cas de valvulopathies rhumatismales et 2 cas d’endocardites infectieuses. L’AVCI par artérite infectieuse représente 8,95% des causes d’AVCI dans notre étude, ce taux est inférieur à ceux des autres séries allant de 17 à 24% des cas (16,27). La recherche étiologique est le plus souvent non concluante dans notre étude du fait de l’inaccessibilité de certaines analyses biologiques ce qui explique que 7 cas d’AVCI dans un contexte infectieux ou post infectieux n’ont pu être documentés. L’infection à VIH était diagnostiquée chez 7 enfants (19), elle constitue une étiologie ou un terrain propice de survenu d’AVCI par vascularite infectieuse. L’AVCH était causé le plus souvent dans notre étude par une rupture d’anévrismes (9 cas), de malformations artério-veineuses (5 cas), ces résultats sont différents des données de la littérature où la MAV était l’étiologie la plus fréquente d’AVCH entre 42,8 et 46% des cas (15,17). Les troubles hématologiques constituent une cause non négligeable d’AVCH, Giroud en 1995 (4) avait trouvé 18% de coagulopathies. La thrombocytopénie est de loin la plus fréquente des hémopathies incriminées dans la survenue des AVCH, elle est reportée entre 30% et 45% dans les séries de Lori,2007(15) et Psaila B,2009 (23). Dans notre étude nous n’avons rapporté qu’un cas de thrombopénie. Nous notons que 43,84% des cas d’AVCH n’avaient pas d’étiologie dans notre série ce qui est similaire aux données de la littérature entre 28,5% et 42,85% (8,17). CONCLUSION : Les AVC de l’enfant demeurent fréquents avec des facteurs de risque modifiables. La fréquence de la drépanocytose et des malformations vasculaires témoigne de la nécessité d’une collaboration multidisciplinaire pour une meilleure prise en charge des enfants.
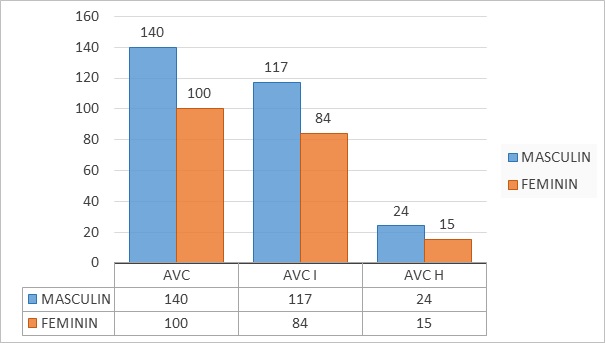 Figure 1e : Répartition selon le sexe des enfants ayant fait un AVC
Tableau 1 : Répartition selon l’âge des enfants ayant fait un AVC
Tableau 2 : Manifestations cliniques des AVC
Tableau 3 : Topographie de l’infarctus cérébral à l’imagerie
Tableau 4 : Topographie de l’hémorragie cérébrale
Tableau 5 : Etiologies des AVCH
Tableau 6 : Facteurs étiologiques des AVCI
Tableau 7 : Cardiopathies emboligènes
LA MYASTHENIE DE L’ENFANT : A PROPOS DE 5 CAS OBSERVES A ABIDJAN EN COTE D’IVOIRE INTRODUCTION : La myasthénie est une maladie auto-immune de la jonction neuromusculaire. Son évolution est souvent capricieuse, marquée fréquemment par des poussées alternant avec des phases de rémission. Les poussées peuvent être provoquées par certains facteurs déclenchants. L’atteinte des muscles respiratoires en fait toute la gravité. La prise en charge précoce et parfois multidisciplinaire et la palette des traitements actuels ont considérablement réduit la mortalité de cette affection avec l’utilisation des immunoglobulines et les échanges plasmatiques (2). La myasthénie de l’enfant est rare (13). Elle représente 10 à 15% des cas de myasthénie chez les caucasiens (15). En Asie particulièrement en Chine, cette fréquence peut atteindre 50% dans la présentation oculaire (20). En Afrique subsaharienne peu de données sont disponibles sur la myasthénie de l’enfant. Deux cas ont été rapportés au Togo (14) et 9 cas au Sénégal (1). En côte d’Ivoire, aucune étude n’a concerné la myasthénie de l’enfant. Notre objectif était de décrire à partir de 5 observations le tableau clinique, et l’évolution sous traitement de la myasthénie chez l’enfant et faire une revue de la littérature sur le traitement. MATERIEL ET METHODES : Nous avons réalisé une étude rétrospective, dans les unités de consultation de neuropédiatrie du CHU de Yopougon et de l’hôpital Mère-Enfant de Bingerville, dans l’unité de consultation du service d’ophtalmologie du CHU de Treichville à Abidjan. Tous les dossiers d’enfants âgés de 0 à 15 ans, chez qui le diagnostic de myasthénie a été posé sur des signes cliniques et/ou ENMG et sur les dosages d’anticorps spécifiques ont été colligés, durant une période de 3 ans, d’Octobre 2015 à Décembre 2018. OBSERVATIONS Observation n°1 : K.M, de sexe masculin, âgé de 14 mois, a été reçu en ophtalmologie en 2015 pour un ptosis bilatéral installé depuis un mois auparavant, d’abord à droite puis à gauche avec un strabisme divergent de l’œil gauche. L’examen physique notait un bon état général, un ptosis bilatéral asymétrique prédominant à droite, maximal en fin de journée. Le test au glaçon était positif. Les tests pharmacologiques n’ont pas été réalisés. Le dosage du taux des anticorps anti-récepteurs à l’Ach et le scanner thoracique n’ont pas été fait. L’ENMG a montré un décrément significatif (fig 1). Le patient a été traité par le chlorure d’ambénonium (Mytelase®). L’évolution a été marquée par une généralisation de la myasthénie, suivie du décès du patient dans un contexte de détresse respiratoire malgré l’intubation et la ventilation en réanimation, 13 mois après la consultation. Le traitement par les immunoglobulines n’a pu être réalisé. Observation n°2 : B.F., de sexe féminin, âgée de 02 ans et 07 mois, a été reçue en neuropédiatrie en 2018, pour un ptosis unilatéral gauche évoluant depuis un mois avec des chutes fréquentes. Ses antécédents personnels étaient marqués par un retard de langage, une non-acquisition de la propreté sphinctérienne et une alimentation faite uniquement de liquides. Elle n’avait pas d’antécédent familial particulier. L’examen physique notait un bon état général, un ptosis unilatéral maximal en fin de journée. Le test au glaçon était positif. Le reste de l’examen neurologique était sans particularité. Les explorations paracliniques ont mis en évidence : un décrément de 10% à l’ENMG, un taux élevé d’Ac anti-récepteurs à l’Ach et une hyperplasie thymique sans processus tumoral individualisable, prédominant à droite au scanner cervico-thoracique (Fig 2). La thymectomie proposée a été refusée par la famille. Sous Mytelase®, l’évolution a été marquée par des épisodes de poussées motivant l’augmentation de la dose de Mytelase et l’association à la corticothérapie. Observation n°3 : K.G, de sexe masculin, âgé de 06 ans, sans antécédent particulier, a été adressé à la consultation de neuropédiatrie en 2017 pour un ptosis fluctuant à bascule, une paralysie de l’abduction de l’œil droit et une chute de la tête évoluant depuis deux mois. Des occlusions oculaires par pansements ont initialement été réalisées sans succès. Secondairement, est apparu le ptosis gauche s’aggravant dans la journée. Le tonus musculaire était normal. Il n’y avait pas d’atteinte d’autres groupes musculaires. Le test au glaçon et les épreuves pharmacologiques n’ont pas été réalisés. L’ENMG objectivait un décrément significatif et le dosage des AC anti-récepteur à l’Ach était positif avec un taux égal à 0,4 nmol/l. Le patient a été traité par la Pyridostigmine ou Mestinon® avec une évolution stationnaire. Observation n°4 : K.Y., sexe masculin, âgé de 08 ans, reçu en 2017, a présenté plus d’un mois avant sa consultation en neuropédiatrie, une fatigabilité à la marche avec réduction du périmètre de marche, sans douleur ; un ptosis fluctuant, une anorexie, une voix nasillarde et une perte de poids d’environ 3 kg en moins d’un mois. Aucun antécédent familial n’a pu être noté. L’examen physique notait un bon état général, un ptosis bilatéral, le test au glaçon et les épreuves pharmacologiques n’ont pas été réalisés. Un décrément significatif a été objectivé par l’ENMG. Le bilan biologique thyroïdien et le scanner thoracique étaient normaux. Le dosage des Ac AntiAch n’a pu être réalisé. Sous chlorure d’ambénonium (Mytelase®) associé à la corticothérapie, l’évolution a été d’abord marquée par une amélioration de la symptomatologie, puis par le décès dans un contexte de généralisation de la myasthénie puis une détresse respiratoire malgré l’intubation et la ventilation en réanimation. Le traitement par les immunoglobulines et/ou l’échange plasmatique n’a pu être réalisé. Observation n°5 : T.M., de sexe féminin, âgée de 12 ans et 09 mois, sans antécédent personnel et familial particulier, a consulté en neuropédiatrie en 2018 pour un ptosis droit invariable au cours de la journée, parfois présent dès le matin au réveil. Une tarsorraphie a été réalisée sans succès. Depuis 5 ans, un ptosis gauche est apparu. Le test au glaçon était positif ; les tests pharmacologiques n’ont pas été réalisés et le dosage des Ac antiAch R était positif avec un taux de 10,8 nmol/L. La patiente a été traitée par la Pyridostigmine (Mestinon®) et l’Azathioprine (Imurel® 25 mg). L’évolution sous traitement a été satisfaisante. DISCUSSION Nous avons recensé 05 cas de myasthénie de l’enfant sur une période de 3 ans. Au Sénégal, sur une période de 5 ans, Basse et al. ont recensé 09 cas de myasthénie chez l’enfant (1). Au Brésil, Morita et al. avaient observé 18 cas de myasthénie de l’enfant sur une période de 14 ans (12). Malgré nos biais de recrutement, toutes ces données semblent confirmer la rareté de la myasthénie chez l’enfant. En Asie, la myasthénie de l’enfant est plus fréquente (20). En Chine, sur une population de 391 myasthéniques, Xiaofan et al ont trouvé 50% d’enfants (19). Aucune donnée de la littérature n’explique clairement cette rareté de la myasthénie de l’enfant. L’âge de nos patients, lors du diagnostic, variait de 01 à 12 ans avec un âge moyen qui était de 5,8 ans. La série sénégalaise situait l’âge moyen de leurs patients au moment du diagnostic à 6,2 ans (1). Vander Pluym et al. ont trouvé un âge de début des symptômes qui se situait entre 18 mois et 11 ans et avant 3 ans dans 8/18 des cas) (18). Deux (2) enfants étaient de sexe féminin, et 3 de sexe masculin. Selon Finnis, à l’âge pré-pubertaire, il y a une égalité au niveau des deux sexes dans la survenue de la myasthénie (6). Comme dans les séries togolaise (14) et sénégalaise (1), les signes inauguraux étaient oculaires représentés par un ptosis sans diplopie. Un seul patient avait des signes bulbaires et spinaux associés au ptosis. Selon Grob et al., classiquement, tous âges confondus, la symptomatologie initiale de la myasthénie est une atteinte oculaire dans 60 % des cas (9). Cette assertion est aussi vérifiée chez l’adulte, comme le démontre l’étude de Gnonlonfoun et al. qui a rapporté un ptosis et/ou une diplopie chez tous leurs patients (8). Le ptosis étant quasi-présent dans les myasthénies juvéniles, il serait souhaitable de réaliser le test au glaçon devant tout ptosis, même si sa positivité ne traduit pas exclusivement la présence d’une myasthénie. Un trouble de la phonation associé à une atteinte oculaire a été retrouvé chez un seul patient. Une atteinte de la musculature spinale, se manifestant par des troubles de la marche ou des difficultés à tenir la tête, était présente chez un seul patient au début de la maladie. Selon Grob et al., 20 % des myasthénies débutent par une atteinte bulbaire ou faciale et 20% des myasthénies débutent par un déficit de la musculature axiale ou périphérique (9). Le délai diagnostique moyen était de 13 mois, avec des extrêmes de 01mois et de 5 ans. Nidain et al. ont rapporté des délais diagnostiques de 6 et 9 ans chez deux sœurs au Togo (14) Ce long délai pourrait s’expliquer par la méconnaissance de la maladie, d’où une errance diagnostique chez différents professionnels de la santé avec un retard au diagnostic ; au cours de cette errance l’une de nos patientes avait même bénéficié d’une tarsorraphie sans succès. Le test au glaçon a été réalisé chez 3 patients sur 5. L’électroneuromyographie (ENMG) avec la technique des stimulations répétitives à basse fréquence de 3Hz en objectivant un décrément significatif a été contributif au diagnostic. L’étude du jitter lors de l’examen en fibre unique n’est pas utilisée en routine (5). Dans notre contexte, le dosage des anticorps est encore très souvent inaccessible dans les hôpitaux publics. Nous avons pu mettre en évidence des anticorps anti-RAch chez 3 patients (60%). La série sénégalaise a retrouvé des Ac anti-RAch dans 77,77% des cas (1). Pour Xiaofan et al., 66% des adultes et 64% des enfants atteints de myasthénie avaient des anticorps anti-RAch (19). Sur nos cinq patients, deux (40%) avaient réalisé un scanner thoracique, l’hyperplasie thymique a été mis en évidence chez un seul. Dans la série du Sénégal, un seul patient avait une atteinte thymique (11,11%) (1). En France, Boumendil et al ont observé une atteinte thymique dans 15 à 20% des cas (3), alors qu’aucune anomalie thymique n’a été rapportée par Gorafalo et al. à Cuba (7). Un thymome est retrouvé chez 10 à 15% des patients atteints de myasthénie avec un pic à l’âge de 50 ans et quel que soit le sexe (17). Les thymomes sont très rares, mais existent dès l’âge de 4 ans. La thymectomie semble améliorer le taux de rémission (4). Parmi les autres affections auto-immunes associées à la myasthénie et qui doivent être recherchées systématiquement, les dysthyroïdies (maladie de Basedow, thyroïdites) sont les plus fréquentes et sont retrouvées chez 5 à 10% des patients (16). Nous n’avons pas objectivé d’affection auto-immune associée à la myasthénie. Par contre, chez l’adulte, Gnonlonfoun et al. ont mis en évidence une hyperthyroïdie chez un de leurs patients (16,67%) (8). Tous nos patients étaient traités par des anticholinestérasiques, 40% par des corticoïdes, et 20% par l’azathioprine. Aucun patient n’a bénéficié d’un traitement par les immunoglobulines intraveineuses ni par du rituximab en raison du coût prohibitif dans notre contexte de travail. Dans la série sénégalaise tous les patients étaient sous anticholinestérasiques et seulement 22,22% sous corticoïdes dès le début de la maladie (1). Dans la série brésilienne, tous les patients ont été traités en première intention par la pyridostigmine. Et pour les mêmes auteurs, le traitement de la myasthénie chez l’enfant se fait selon la sévérité de l’atteinte, à la recherche d’une rémission complète des symptômes (12). Selon la littérature, les anticholinestérasiques sont utilisés en première intention (10). S’ils sont efficaces dans 50 à 70% des cas sur un ptosis isolé, ils restent le plus souvent insuffisants lorsque des troubles oculomoteurs sont présents, et ne dépassant pas 20% d’efficacité. Chez ces patients, non répondeurs aux anticholinestérasiques, les immunosuppresseurs sont plus efficaces. L’azathioprine reste le premier choix pour un traitement immunosuppresseur à long terme (11). La corticothérapie est généralement utilisée lorsque les symptômes ne sont pas suffisamment contrôlés par les anticholinestérasiques, Les résultats sont plutôt bons, puisque chez 72 à 92 % des patients traités, elle a entrainé une rémission ou une amélioration importante (17). Rarement utilisé seul du fait de son long délai d’action, l’azathioprine est actuellement souvent utilisée en association avec les corticoïdes (17) avec de très bons résultats. Cette association permettrait de consolider une rémission mais également de réduire la posologie des corticoïdes limitant ainsi leurs effets secondaires (5). Les immunoglobulines et les échanges plasmatiques, le rituximab sont actuellement disponibles dans notre contexte. Mais l’accès aux immunoglobulines ou aux échanges plasmatiques ou au ritixumab reste encore difficile. L’indication de ces médicaments suscités a été posé chez deux patients, le coût a été un frein. L’évolution sous traitement était marquée par une amélioration chez deux de nos patients. Chez un patient, nous avons noté une aggravation qui a motivé l’augmentation des doses d’anticholinestérasiques. Le décès est survenu chez deux autres patients après généralisation de la myasthénie et dans un contexte de détresse respiratoire, malgré l’association d’une corticothérapie. Il apparait donc nécessaire de surveiller les formes oculaires chez l’enfant même s’il est admis que la myasthénie du sujet âgé semble se généraliser plus fréquemment que chez le sujet jeune (5). La série sénégalaise a noté une amélioration nette chez 4 patients sur 9 et la survenue de 2 décès dans un tableau d’insuffisance respiratoire aigüe (1). Au Togo, les patientes ont été perdus de vue (5). Sur tous les patients de Morita traités à la pyridostigmine, il y a eu une rémission complète chez deux patients (12,5%) et trois patients (18,7%) ont présenté une amélioration clinique ; l’association de la prednisone chez 9 patients n’a pas entrainé de rémission complète, mais une stabilité chez 5 patients (50%) (12). En Chine, plusieurs enfants ont reçu de la prednisone sans rémission clinique évidente (19). Les formes oculaires de la myasthénie sont l’apanage de l’enfant à l’âge pré-pubertaire, avec une évolution vers la myasthénie généralisée à l’âge pubertaire. La rémission spontanée ou après traitement s’observe beaucoup plus à l’âge pré-pubertaire (6). CONCLUSION L’analyse des 5 observations de myasthénie chez l’enfant, a montré qu’elle a un début oculaire (ptosis), dans la quasi-totalité des cas. L’errance diagnostique est fréquente dans notre contexte de travail liée à la méconnaissance de l’affection. Il convient donc d’évoquer la myasthénie devant tout ptosis uni ou bilatéral, asymétrique et/ou fluctuant, chez l’enfant et de réaliser un test au glaçon, ainsi que, dans la mesure du possible, les tests aux anticholinestérasiques et le dosage des Ac anti-RAch. La séropositivité d’anticorps anti-récepteurs à l’acétylcholine est fréquente et souvent associée à une tumeur de thymus à rechercher obligatoirement. L’évolution est comparable à celle de l’adulte car la généralisation suivie du décès n’est pas rare. Le traitement est basé sur les anticholinestérasiques qu’il faut associer à la corticothérapie et/ou à l’azathioprine en cas de non réponse aux anticholinergiques. La plasmaphérèse, les Immunoglobulines et le rituximab ont révolutionné le pronostic de cette maladie, mais ces moyens thérapeutiques restent inaccessibles dans notre contexte en raison de leur coût. Une subvention de ces médicaments par nos états pourrait améliorer considérablement la prise en charge.
 Figure 1: Décrément à l’ENMG
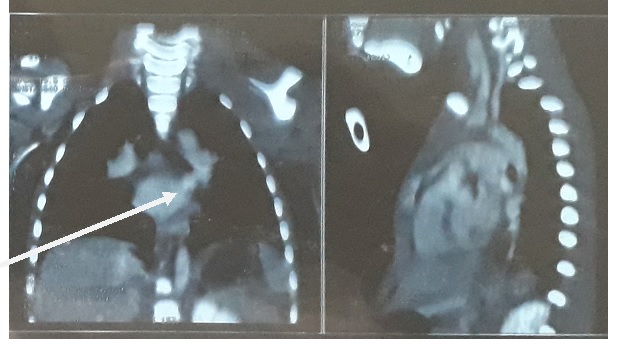 Figure 2: hyperplasie thymique au scanner cervico-thoracique.
Tableau I : Répartition des patients en fonction de l’âge et du sexe
Tableau II : Manifestations cliniques initiales et évolution sous traitement
ETIOLOGIES DES HYDROCEPHALIES DE L’ENFANT AU CENTRE HOSPITALIER UNIVERSITAIRE YALGADO OUEDRAOGO (CHU-YO) DU BURKINA FASO INTRODUCTION L’hydrocéphalie se définit comme un trouble de l’hydrodynamique du LCS, entrainant une distension progressive des cavités ventriculaires cérébrales et parfois des citernes et des espaces sous-arachnoïdiens péri-cérébraux en rapport avec une hyperpression du LCS. C’est une pathologie qui est souvent responsable de séquelles neuropsychiques pouvant engager le pronostic vital. Les principales étiologies sont regroupées en causes malformatives, infectieuses, tumorales et vasculaires/hémorragiques. En Afrique sub-saharienne, on enregistre plus de 200000 nouveaux cas d’hydrocéphalie par an chez les enfants et l’origine infectieuse prédomine de façon générale (11). Une étude réalisée en 2010 au CHU-YO de Ouagadougou avait retrouvé une prédominance des causes infectieuses (43,4%) des hydrocéphalies chez l’enfant (12). Après une décennie, nous nous proposons d’actualiser le profil étiologique des hydrocéphalies de l’enfant au CHU-YO. PATIENTS ET METHODE L’étude s’est déroulée dans le service de Neurochirurgie du centre hospitalier Universitaire Yalgado OUEDRAOGO (CHU-YO) au Burkina Faso. Il s’agissait d’une étude rétrospective descriptive et transversale sur une période de 2 ans allant du 1er janvier 2018 et le 31 décembre 2019. Etaient concernés par l’étude, les enfants de 0 à 15 ans hospitalisés dans le service pour hydrocéphalie confirmée par au moins un scanner cérébral. Les patients n’ayant pas bénéficié d’une prise en charge chirurgicale ont été exclus. Les données épidémiologiques, les antécédents obstétricaux et médicaux, les signes d’examen physique et les résultats d’imagerie étaient les variables étudiées. RESULTATS Quatre-vingt-dix-sept (97) cas d’hydrocéphalies infantiles répondaient à nos critères d’inclusions et ont été admis pour l’étude. L’âge moyen à l’admission était de 33,6 mois avec des extrêmes de 1 et 180 mois. On notait un sex ratio de 0,7. Les nouveau-nés et les nourrissons représentaient 68% de notre échantillon. Le délai moyen de consultation était de 4,76 mois. Les principaux signes cliniques étaient la macrocranie (73,2%) avec un périmètre crânien moyen de 51,7 cm, le bombement de la fontanelle antérieure (42,3%), le syndrome d’hypertension intracrânienne (29,9%), le signe de Parinaud (20,7%) et le retard psychomoteur (16,5%). La tomodensitométrie cérébrale a été réalisée chez tous les patients. Huit patients ont pu bénéficier en plus, d’une imagerie par résonance magnétique afin d’affiner le diagnostic étiologique et de proposer une meilleure stratégie thérapeutique. L’hydrocéphalie était non communicante dans 57,7% des cas et communicante dans 42,3%. L’étiologie était malformative (n=60 soit 61,9%), tumorale (n=15 soit 15,5%), infectieuse (n=3 soit 3,1 %) et vasculaire (n=2 soit 2%). L’étiologie de l’hydrocéphalie était indéterminée dans 17,5% (n=17) des cas. Le Tableau 1 présente la répartition des patients selon l’étiologie de l’hydrocéphalie. Les malformations associées à l’hydrocéphalie (n=60) étaient représentées par la myeloméningocèle (31,7%), la sténose congénitale de l’aqueduc du mésencéphale (31,7%), la malformation de Dandy-Walker (28,4%), le kyste arachnoïdien intracrânien (5%), la céphalocèle (1,6%) et l’atrésie congénitale des foramens de Monro (1,6%). La figure 1 illustre une malformation de Dandy-Walker sur le plan scanographique et le tableau 2 donne la répartition des hydrocéphalies d’origine malformative. Concernant les étiologies tumorales (n=15), la tumeur était localisée au niveau de la fosse crânienne postérieure dans 53,3% des cas et en sus-tentoriel dans 46,7% où elles siégeaient exclusivement sur la ligne médiane composés et comme suit : région suprasellaire (33,3%), région pinéale (6,7%) et troisième ventricule (6,7%). La figure 2 illustre une hydrocéphalie d’étiologie tumorale. Les étiologies infectieuses étaient représentées par 3 cas d’hydrocéphalie avec antécédent d’hospitalisation en pédiatrie pour une méningite bactérienne devant le tableau clinique et l’aspect du liquide cérébro-spinal très évocateurs. Les germes impliqués n’avaient pas été mis en évidence par la bactériologie. Concernant les étiologies vasculaires/hémorragiques (n=2), il s’agissait d’un cas de malformation artério-veineuse et un cas d’hémorragie sous-arachnoïdienne post-traumatique. Pour la prise en charge chirurgicale, la dérivation venticulo-peritonéale était réalisée dans 76,3% des cas. Elle était associée à la cure d’une myeloméningocèle dans 14,9% des cas. La ventriculo-cisternostomie endoscopique avait été pratiquée chez 20,7% des patients. Les autres traitements étaient : dérivation ventriculaire externe (1%), dérivation kysto-péritonéale (1%), marsipuilisation du kyste arachnoïdien dans le ventricule latéral (1%). Parmi les patients qui présentaient des tumeurs (n=15 soit 15,5%), l’abord chirurgicale de la lésion tumorale a été pratiqué dans 40% (n=6) des cas en plus de la dérivation de LCS: les diagnostics anatomo-pathologiques étaient les suivants : astrocytome pilocytique, astrocytome sous épendymaire à cellules géantes, craniopharyngiome, médulloblastome, hémangioblastome, pinéaloblastome. Dans notre série (n=97), l’évolution post-opératoire à moyen terme (3 à 6 mois) était simple dans 72,2% des cas et compliquée dans.22,7%. Le décès était survenu dans 5,1% des patients. DISCUSSION Notre étude révèle une nette prédominance des étiologies malformatives dans les hydrocéphalies chez les enfants de 0 à 15 ans pris en charge dans notre service. Ces résultats constituent une inversion des tendances épidémiologiques jadis retrouvés aussi bien dans notre pays (12) qu’ailleurs (2,10,11). En effet, les causes infectieuses occupent une part importante dans le profil étiologique des hydrocéphalies en Afrique subsaharienne. En témoignent les études réalisées au Burkina par Tapsoba et al. en 2010 (12) qui avait retrouvé une prédominance infectieuse avec des proportions respectives de 43,4%. Les travaux réalisés dans d’autres pays notamment au Benin (Fatigba en 2010) et au Sénégal (Ba en 2012) avaient retrouvé des résultats similaires avec une prédominance infectieuse dans respectivement 80,7% (10) et 46% (2). Toutefois cette inversion du profil étiologique des hydrocéphalies de l’enfant marquée par la prédominance des causes malformatives qui surpassent celles infectieuses a été également constatée par d’autres auteurs en Afrique. Il s’agit de Broalet et al. en 2011 en côte d’ivoire (5) , Diarral et al. en 2018 (9) au Mali et Dakurah et al. au Ghana en 2016 (7) Cette inversion du profil étiologique de l’hydrocéphalie de l’enfant dans notre contexte pourrait être expliquée d’une part par la baisse des causes infectieuses et d’autre part la persistance de causes malformatives qui sont de mieux en mieux diagnostiquées avec l’arrivée récente du scanner ou mieux de l’imagerie par résonance magnétique dans nos pays. En effet, la baisse des causes infectieuses dans notre service pourrait s’expliquer par un diagnostic et traitement précoce des pathologies infectieuses comme les méningites; rendus possible par la gratuité des soins chez les enfants de moins de 5 ans (Décret du Président du Faso en 2016) et par l’efficacité de la mise en œuvre du programme élargi de vaccination dont la couverture vaccinale pour les vaccins anti-pneumococcique et anti-Haemophilus influenzae B atteignait 100% en 2018 (annuaire statistique 2018, ministère santé du Burkina Faso). Par contre la persistance des causes malformatives pourrait s’expliquer par le taux faible de suivi des grossesses et le manque de prévention des malformations du système nerveux central. Selon l’annuaire statistique 2018 du ministère de la santé du Burkina, seulement 37,8% des femmes enceintes avaient bénéficié d’une consultation prénatale (CPN) au 1er trimestre de leur grossesse. Les CPN étant une occasion pour supplémenter les femmes enceintes en acide folique. Du reste, cette supplémentation au cours de la grossesse demeure insuffisante car elle devrait commencer bien avant la conception. Plusieurs études (1,3,4,8) ont démontré l’intérêt de la supplémentation pendant la période péri-conceptionnelle en acide folique dans la prévention des anomalies de fermeture du tube neural qui constituent une part importante des associations lésionnelles responsables d’hydrocéphalie dans notre étude. Les causes tumorales représentaient 15,5% des étiologies d’hydrocéphalie avec une prédominance des tumeurs de la fosse crânienne postérieure (53,3%). Ces résultats corroborent ceux de la littérature. En effet, Togo et al. en 2019 (13) au Mali trouvaient que la localisation sous-tentorielle représentait 83% des tumeurs de l’enfant. En outre, selon Caire et al. en 2015, les tumeurs représentaient 20% des causes d’hydrocéphalie de l’enfant et étaient dominées par les tumeurs de la fosse cérébrale postérieure et de la ligne médiane: médulloblastome, tumeurs de la région pinéale, astrocytome, épendymome, craniopharyngiome (6). Dans notre étude, seulement 40% patients porteurs d’une tumeur avaient bénéficié d’un abord chirurgical de la tumeur avec étude anatomo-pathologique. Le faible taux de chirurgie des tumeurs retrouvées dans notre contexte s’expliquerait par l’état clinique précaire de certains patients, la localisation profonde de la tumeur, le manque de moyens financiers et parfois l’insuffisance du plateau technique (neuro réanimation, traitement adjuvant à la chirurgie). Les étiologies vasculaires/hémorragiques représentaient 2% des étiologies. Ces résultats pourraient s’expliquer par l’âge de notre population d’étude. En effet, ces étiologies sont fréquentes chez les prématurés avec la rupture des vaisseaux lié à leur fragilité (6,11). Dans les pays développés, l’hémorragie intraventriculaire du prématuré est une étiologie fréquente d’hydrocéphalie de l’enfant (11). L’absence de prématuré dans notre échantillon pourrait expliquer cette proportion d’hydrocéphalie d’origine vasculaire/hémorragique. Dans notre étude, 17,5% des patients avaient une étiologie indéterminée. Il s’agissait des cas d’hydrocéphalie communicante (sans malformation, ni tumeur, ni saignement visualisé a la TDM) sans antécédent d’infection cérébro-méningée ou de traumatisme crânio-encéphalique et chez qui l’examen physique ne retrouvait pas de syndrome infectieux ou méningé. Nous pensons qu’il pourrait s’agir des cas dont le diagnostic étiologique nécessiterait des examens paracliniques comme l’imagerie par résonnance magnétique. Les principales limites de notre étude sont relatives à son caractère rétrospectif et au problème d’archivage des dossiers cliniques qui ne nous ont pas permis de recueillir toutes les informations concernant les antécédents anténataux, per et postnataux de nos patients. CONCLUSION Notre étude montre une prédominance des malformations dans le profil étiologique des hydrocéphalies de l’enfant de 0 à 15 ans. Comparée aux études antérieures du service, il s’agit d’une inversion du profil étiologique des hydrocéphalies de l’enfant marquée par la prédominance des causes malformatives qui surpassent celles infectieuses. Cela s’expliquerait par une efficacité dans la prévention (Programme Elargi de Vaccination), le diagnostic et le traitement des infections cérébro-méningées. Ces résultats suggèrent beaucoup d’efforts dans la surveillance des grossesses et l’adoption des mesures de prévention des malformations du système nerveux central telle que la supplémentation péri-conceptionnelle en acide folique.
Tableau 1 : Répartition des patients selon l’étiologie de l’hydrocéphalie
Tableau 2 : Répartition des patients selon le type de malformation associée à l’hydrocéphalie
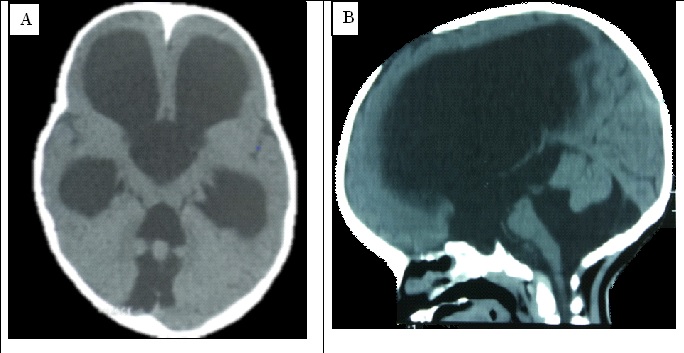 Figure 1 : TDM cérébrale sans injection de produit de contraste en coupe axiale (A) et reconstruction sagittale (B) illustrant une malformation de Dandy-Walker.
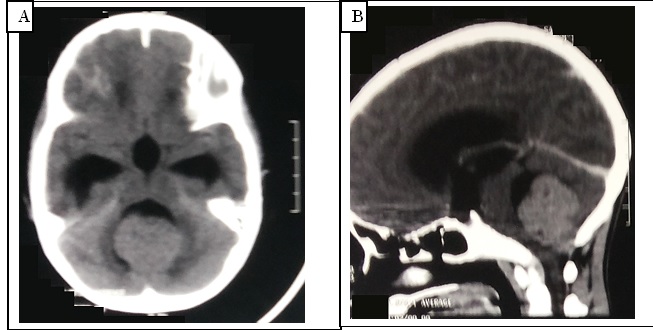 Figure 2: TDM cérébrale en coupe axiale non injectée (A) et reconstruction sagittale après injection de produit de contraste (B) permettant d’objectiver une lésion évocatrice d’une tumeur de la fosse cérébrale postérieure comprimant le quatrième ventricule et entrainant une hydrocéphalie en amont. INTRODUCTION La fréquence des suppurations intracrâniennes reste élevée dans les pays en voie de développement contrairement aux pays développés où elles sont de plus en plus rares. Les enfants sont plus touchés que les adultes. La mortalité et des séquelles restent également importantes (1, 5, 8, 12). Bien que ces affections aient fait l’objet de plusieurs études en Afrique sub saharienne, les études sur le devenir des enfants traités pour ces affections restent quasi inexistantes, notamment celles concernant leur scolarisation. Au Cameroun, leur prise charge s’améliore au fil des années, parallèlement à l’amélioration des moyens diagnostiques et thérapeutiques (6, 13). Le but de cette étude était d’évaluer le pronostic à long terme des enfants opérés des suppurations intracrâniennes et leur scolarisation. METHODOLOGIE : Nous avons mené une étude transversale multicentrique, rétrospective sur 10 ans, du 1er Janvier 2010 au 31 Décembre 2019. Nous avons étudié les aspects diagnostic, thérapeutique, et pronostic, les dossiers des patients de moins de 17 ans, opérés pour une suppuration intracrânienne dans les services et unités de neurochirurgie de l’Hôpital Central et l’Hôpital Général de Yaoundé et de l’Hôpital Général de Douala. Les familles et/ou les patients ont été contactés pour répondre à un questionnaire par rapport à leur évolution scolaire. RESULTATS : Au total, 33 patients ont été inclus dans cette étude dont 24 garçons (72,7%) et 9 filles (27,3%). L’âge moyen était de 9,75±4,59 ans (extrêmes 3 mois et 16 ans), 6 enfants (18,1%) avaient un âge compris entre 0 et 5 ans. Au moment de la survenue de la maladie, 27 patients (81,9%) étaient scolarisés, 6 (18,1%) non scolarisés et 30 (90%) résidaient en zone urbaine. Le délai moyen de consultation était de 11,39±4,49 jours. Les infections ORL constituaient le foyer primaire dans 48,5% des cas. Les signes cliniques fréquents étaient la fièvre 31 cas soit (93,9%), les céphalées 24 cas soit (72,7%), l’altération de la conscience 19 cas soit (57,6%) et les convulsions 12 cas (36,4%). L’altération de la conscience était évaluée avec le Score de Coma de Glasgow (SCG) (14-13) chez 10 patients (52,63%), un SCG (12-9) chez 07 patients (36,84%) et un SCG (≤ 8) chez 02 des patients (6,1 %). La triade de Bergman était présente chez 13 patients (39,4%). Les cultures étaient positives chez 13 patients (39,4%) et le germe le plus retrouvé était le Staphylococcus aureus 6 cas (41,1%) et le Streptococcus sp, 4 cas (30,4%). Les différentes formes anatomiques se répartissaient ainsi qu’il suit : les empyèmes 17 cas (51,5%), les abcès 13 cas (39,4%), la pyoventriculite un cas (0,3%), l’association abcès et empyème deux cas (6,1%). Sur le plan chirurgical, la trépano-ponction avec drainage et lavage de la suppuration au sérum physiologique a été pratiquée dans neuf cas d’abcès (69,2%) et six cas d’empyèmes (35,3%), tandis que la craniotomie l’a été dans quatre cas (30,8 %) d’abcès et 11 cas (64,7%) d’empyèmes. Le cas de pyoventriculite et les deux cas d’empyèmes associés à l’abcès ont bénéficié d’une craniotomie. Concernant la prise en charge médicale tous les patients ont reçu une triple antiobiothérapie probabiliste (Céphalosporine de troisième génération, Aminoside et métronidazole), adaptée à l’antibiogramme par la suite. La durée d’hospitalisation moyenne était de 14,12±8,38 jours. Il y a eu 5 (15,15%) cas de décès au total, 2 cas au cours de la première année post opératoire et 3 cas au cours de la deuxième année post opératoire. Nous n’avons observé aucun cas de récidive de suppuration pendant la période d’étude. L’évolution des patients à un an post opératoire est présentée au tableau I. L’évolution et profil de la scolarisation à la fin de l’étude sont présentés au tableau II. DISCUSSION Ce travail, premier de son genre dans son volet abordant la rescolarisation des enfants opérés de suppurations intracrâniennes dans notre milieu a eu pour principale difficulté, la disponibilité des familles à l’endroit de l’équipe chargée d’évaluer les patients opérés plusieurs années auparavant. Mais cela n’a pas entaché la poursuite de l’étude jusqu’à son terme. Ainsi cette étude a montré que la tranche d’âge la plus représentée était comprise entre 11 et 15 ans. Cette caractéristique était aussi rapportée par d’autres séries africaines notamment en Côte d’ivoire (3) et au Sénégal (1). Ces pathologies touchent donc plus des enfants déjà scolarisés. Comme dans plusieurs études antérieures, les enfants de sexe masculin sont les plus affectés et les infections ORL représentent la principale porte d’entrée (1,6,12). L’identification de la porte d’entrée a permis une meilleure prise en charge du fait que son éradication limite au strict minimum la possibilité de récidive. L’altération de la conscience était retrouvée dans 57,6% et les convulsions dans 36,4% des cas. Ces symptômes, en raison de leur impact sur un cerveau en croissance et maturation doublée de l’agression chirurgicale comme c’est le cas pour notre population d’étude sont susceptibles d’entraver la poursuite normale des études. Cette étude a retrouvé un faible nombre de cultures positives parmi les suppurations analysées. Ce résultat pourrait s’expliquer par le fait que le diagnostic des suppurations intra crâniennes est généralement tardif dans note milieu et les patients de ce fait sont référés tardivement vers les centres spécialisés. En conséquence, la plupart des patients avant le transfert, sont traités par des antibiotiques de façon empirique et probabiliste avec un risque de décapiter l’infection. Cependant, dans cette étude, les enfants opérés ont retrouvé une scolarisation quasi normale pour une large majorité d’entre eux. Au Cameroun, les services de Neurochirurgie ne disposent pas encore de stéréotaxie ou de neuronavigation. En conséquence, les deux techniques chirurgicales pratiquées dans cette étude étaient la trépano-ponction et la crâniotomie au cas par cas. Ces techniques sont largement partagées par la littérature pour ces pathologies (1,2,4,9,11). L’amélioration du plateau technique a grandement impacté le bon suivi et le devenir des patients en Afrique. A la fin de l’étude le taux de mortalité était de 9,7% et la principale séquelle était l’épilepsie (22,6%). Ce résultat nous a paru satisfaisant dans notre cadre d’exercice, considérant le fait qu’il n’y avait pas d’étude antérieure sur le pronostic à long terme au Cameroun sur ces pathologies. L’épilepsie est une manifestation connue de ces pathologies et/ou de leurs séquelles (7,10). Dans cette étude, malgré les séquelles, les enfants opérés ont retrouvé une scolarisation quasi normale dont 46,4% d’entre eux étaient dans l’enseignement supérieur. CONCLUSION : Les suppurations intracrâniennes affectent plus le grand enfant et l’adolescent dans notre milieu. Les infections ORL représentent le principal foyer primaire. Le pronostic à long terme reste satisfaisant avec l’épilepsie comme principale séquelle. La scolarisation de ces enfants reste quasi normale en cas de guérison sans séquelle, mais tous les survivants ont pu reprendre leurs études.
Tableau I : Évolution clinique à un an post-opératoire
Tableau II : Evolution clinique et profil de la scolarisation à la fin de l’étude (N = 28).
GLIOMA GENETIC SUSCEPTIBILITY AND SURVIVAL ANALYSIS IN THE EAST ALGERIAN POPULATION INTRODUCTION Gliomas are the largest group of Central Nervous System (CNS) tumors. Compared to the other cancers, they are relatively rare with a poor prognosis. They represent 1.6% of new cancers and 2.5% of cancer deaths worldwide (8). Genetic factors have been validated as important contributors for these neoplasms and recent genome-wide association studies (GWAS) have demonstrated that the inherited risk is due to the coinheritance of multiple low-risk genetic variants. So far, 25 risk loci have been identified as influencing risk (17,20,25). The GWA studies were carried on populations of developed countries (European and American). The Chinese population has revealed that not all the reported glioma risk–associated variants identified through GWAS are associated with glioma susceptibility (9,10). Data on developing countries remain very limited. That is the case of Algeria, where the genetic structure is highly heterogeneous (5) and the knowledge about gliomas remain scarce. Very few studies have been done and the limited budgets devoted to research in this developing country is one of the most limiting factors. Moreover, the lack of biological sample collection structure has driven researchers to focus much more on the most prevalent diseases. The aim of this study was to investigate the glioma risk and the survival of patients in the east Algerian population based on the polymorphisms identified by GWASs. SUBJECTS AND METHODS 2.1 Studied population This case-control study involved 76 diffuse glioma patients and 82 apparently healthy individuals. Cases were recruited at the University Hospital Benbadis Constantine, where the majority of cancer patients from Eastern Algeria are received. Between March 2014 and October 2016, the researcher asked the patients about their willingness to participate in the study. Cases and controls were frequency matched on age and sex and they were from the same geographic region. The demographic and personal data were collected via a structured questionnaire. The clinical characteristics of the cases were obtained from medical records. Histopathological classification was based on the World Health Organization 2007 criteria (18). All the participants had no previous history of cancer and CNS-related diseases. Written informed consent was obtained from each study subject prior to their participation in the study. The use of human blood sample and the protocol in this study strictly conformed to the principles expressed in the Declaration of Helsinki. 2.2 DNA preparation and SNP genotyping Genomic DNA was extracted from peripheral blood samples using a standard salting out method (22). DNA concentration and purity were evaluated using a spectrophotometer (NanoDrop 2000; Thermo Fisher Scientific, Waltham, MA, USA) and the PicoGreen technique specific to double strand DNA. Seventeen of the Single Nucleotide Polymorphisms (SNPs) identified by GWASs with a Minor Allele Frequency (MAF) of at least 0.2 were selected for genotyping. Table 1 shows our selection of SNPs and their basic characteristics. Genotyping was performed on the Sequenom MassARRAY platform (Sequenom, San Diego, CA, USA). Primers for polymerase chain reaction (PCR) amplification and single-base extension assays were designed using the Assay Design Suite V2.0 (Agena BioscienceTM). The reactions were performed using the iPLEXTM Gold reagent kit. Data were collected with the Mass ARRAY System, a MALDI-TOF (Matrix-Assisted Laser Desorption Ionization – Time of Flight) mass spectrometer, and genotypes were analyzed using Sequenom Typer 4.0 Software. For quality control, 5% of samples were randomly selected, and the results showed 100% concordance. 2.3 Statistical analysis Continuous variables were analyzed by Student t test and categorical variables by χ2-test. The Hardy-Weinberg equilibrium (HWE) was assessed for each SNP in controls. The correlation between polymorphisms and glioma development was estimated by Fisher’s Exact test and the results were expressed by Odds ratio (OR) and 95% confidence interval (95% CI). Under an additive model, the Wilcoxon rank sum test was used to analyze the association between the total number of risk alleles and glioma susceptibility. For survival analysis, the Kaplan-Meier method was used. The overall survival (OS) was defined as the time lapse from the day after surgery to the date of death or last contact. Univariate and multivariate analysis using the Log-rank test and the Cox proportional hazard model were performed to identify prognosis factors. P < 0.05 was considered as statistically significant. All analyses were conducted using R software (R-3.4.3). RESULTS Our analysis included 65 of our diffuse glioma patients and 72 of controls who were satisfactorily genotyped. The 17 SNPs tested were within HWE in controls. 3.1 Studied population The demographic and clinical characteristics of patients and controls are reported in Table 2. Cases and controls were matched for age and sex, and there was no significant difference between them (p=0.64 and 0.78 respectively). The glioma group had a mean age of 50.3 years and was 66% male. Five of the patients had a low-grade glioma (grade II) and 60 had a high-grade tumor (grade III, n = 13; grade IV, n = 47). Glioblastoma was the most frequent histological subtype with 46 cases. Frequencies of the other types were as follows: astrocytoma (n = 5), oligodendroglioma (n = 6), oligoastrocytoma (n = 3), ependymoma (n = 3), gliosarcoma (n = 1) and glioma Not Otherwise Specified (n = 1). For statistical analysis, tumors were distributed as GBM or non-GBM. The gliosarcoma (grade IV) was integrated with the GBM group. 3.2 Case-control analysis Table 3 shows the results of the correlation between our selection of polymorphisms and glioma risk in our cohort. In the analysis of each SNP, the rs2736100 at 5p15.33 in intron 2 of TERT gene was the unique polymorphism that has shown a significant p-value (p = 0.006), but it seems to have a protective effect (OR = 0.21; 95%CI [0.06-0.64]). The total number of risk alleles was not associated with the risk of glioma (p=0.38). After Bonferroni correction for multiple comparisons, the rs2736100 was no longer significant (p = 0.25). 3.3 Survival analyses We found that patients aged ≥ 50 years had a 2.15-fold increased risk of death on overall survival (OS) (hazard ratio (HR) = 2.15, 95% CI: [1.19 – 3.88], p = 0.01) and that Non-GBM tumors were associated with 74% decrease in mortality hazard (HR = 0.26, 95% CI: [0.12-0.55], p = 0.0003) compared to GBM. The mean survival time of our cohort was 19.6 months, the median 12.2 months and 95% CI: [10.03-18.03]. In the univariate analysis, beside the age (p= 0.006), sex (p= 0.02), glioma subtype (p= 2e-04) and WHO grade (p= 0.001), three SNP were identified as prognosis factor, i.e. rs12076373 (p = 0.03), rs3751667 (p = 0.02) and rs10852606 (p = 0.03). However, in multivariate analysis none of them was confirmed. The total number of risk alleles was not associated with the survival of patients (p = 0.2) neither the rs2736100 (p = 0.8). DISCUSSION The genetic bases of GWAS, that have provided evidence for a polygenic susceptibility to glioma, are European and American (17,29,31,33,34,37,38). Studies on the Chinese population strongly suggested the important genetic heterogeneity in glioma risk (9,10). Within the North African context, the genetic composition of the Algerian population is an amalgam of different ancestral component coming from the middle East, Europe, sub-Saharan Africa and autochthonous to North Africa (Maghrebi) (3-5,13,15). With a such genetic wealth, data on glioma and rare diseases in Algeria and North Africa remain scarce because of the limited resources in biomedical research (16,32,36). To our knowledge, our study is one of the first investigations on glioma susceptibility in the region. The risk of glial tumors in our population was associated with none of the 17 SNPs analyzed neither with the total number of risk alleles. Nonetheless, our results revealed an interesting polymorphism, the rs2736100 (5p15.33, TERT), which has shown a protective effect on glioma (OR = 0.21; 95% CI [0.06-0.64]) before the Bonferroni correction. The risk locus 5p15.33 harboring the TERT gene, encoding the catalytic subunit of telomerase, has been implicated in several kinds of cancer (21,28) and the rs2736100, located in intron 2 of the TERT, has been linked to increased risk of glioma since the two first GWAS (33,38) and confirmed by several studies (40). The absence of association between risk SNPs identified by GWASs and susceptibility to glioma as well as the protective effect of the rs2736100 in our cohort may be explained by ethnicity, since risk-allele frequencies correlate modestly between ancestry groups (23) and the identification of disease-associated SNPs by GWA studies tends to have low concordance when different populations are compared (39). Indeed, a same allele may be a risk factor in a population and a protective factor in another (12,35). Furthermore, many of the disease risk variants discovered by GWAS are shared across Eurasians, while the replication with individuals of African ancestry is much less common (19). The survival analysis has shown that the overall survival in our cohort (mean 19.6 months and median 12.2 months) join other populations all over the world (1,2,7,24) as well as the known prognosis factors of glioma (i.e. age, sex, histological subtype and grade of malignancy) (6,14,24,27,30). Moreover, three SNPs (rs12076373, rs3751667 and rs10852606) may be associated with the survival of patients in our population, while a unique susceptibility variant (rs78378222) was associated with survival in GWAS (11). These associations, even if not confirmed, may indicate that variants identified by GWAS may be prognosis markers in our population. This is supported by the findings of Ostrom et al., who reported differences in incidence and survival of glioma by racial or ethnic groups (26). Our results should be taken with caution since the modest sample size reduces considerably the power to detect all the susceptibility variants and prognosis factors in the Algerian population. CONCLUSION: In summary, our results indicate that germline risk variant of gliomas in our population may be different from those identified by GWASs and that some polymorphisms may be linked to the survival of patients. A larger sample is needed to identify the real glioma risk variants implicated in the Algerian population and sequencing the 5p15.33 region could reveal point mutations specific to this ethnic group. Conflict of interest: The authors declare no conflict of interest. Funding: This research was funded by the university of Constantine 1 Algeria. I, the undersigned Sabrina TOUATI, first author, certify that all the persons cited have read and approved the mention of their name in the article
TABLES: Table 1: Basic information on the 17 Single Nucleotide Polymorphisms (SNPs) analyzed
GBM: Glioblastoma
Table 2: Demographic and clinical characteristics of participants
SD: Standard Deviation
Table 3: Odds ratios, 95% confidence interval and p-values of the 17 tested SNPs
Articles récents
Commentaires récents
Archives
CatégoriesMéta |
© 2002-2018 African Journal of Neurological Sciences.
All rights reserved. Terms of use.
Tous droits réservés. Termes d'Utilisation.
ISSN: 1992-2647